Introduction
Coagulation factor V (FV) is a single-chain glycoprotein that plays an important role in the coagulation cascade as a cofactor of activated FX (FXa) in the prothrombinase complex
[1-3]. Hepatocytes and megakaryocytes synthesize human FV with a molecular weight of 330 KD and about 80% circulates in the blood, while about 20% of total FV is stored in the α-granules of platelets [4, 5].
FV and FVIII show structural and functional homology and are composed of six domains, including A1-A2-B-A3-C1-C2 [6, 7]. Three homologous A-type domains are connected to two homologous C-type domains by a heavy glycosylated B domain. After being released, FV converts to activated FV (FVa) by the proteolysis cleavage of some proteases such as FXa and/or thrombin. These proteases cleave FV at 3 Arginine residues, including R709, R1018, and R1545, and the B domain is removed. The result of this enzymatic cleavage is FVa which consists of a 105-KD heavy chain (A1-A2) and a 71-KD light chain (A3-C1-C2) [4, 8-12].
The gene for human FV (F5) has been mapped to chromosome 1q23, and its length is more than 80 kb. The gene consists of 25 exons [13]. Exon number 1 to 12 are responsible for encoding the heavy chain and the light chain is encoded by exones 14 to 25, while exon 13 encods the B domain [14, 15]. FV messenger RNA encodes a signal peptide of 28 amino acids and a mature peptide of 2196 amino acids [16, 17].
Congenital FV deficiency or parahaemophilia is an autosomal recessive disorder that Owern first described in 1947 and is characterized as a rare bleeding disorder with a frequency of about 1 in 1,000,000 [18, 19]. Mutations in F5 can lead to 2 different types of FV deficiencies: type I or quantitative defect, which is characterized by reduced both FV activity (FV: C) and FV antigen (FV: Ag) level and type II (qualitative) with only reduced FV: C [20]. Severe FV deficiency is characterized by <1% of regular FV activity and very low antigen level [21]. Although cases with life-threatening bleeding such as central nervous system bleeding and gastrointestinal bleeding have been reported in severe factor V deficiency [22-25], patients with severe FV deficiency usually show mild to moderate phenotypes in terms of bleeding symptoms [21]. There is not a strong correction between the level of FV and the severity of bleeding symptoms in FV deficiency, contrary to what is seen in hemophilia A or B. However, patients with lower FV levels are usually more prone to bleeding [26], but some patients with very low factor levels may not experience severe symptoms [24, 27, 28].
In Iran due to the high rate of consanguineous marriages in some areas, autosomal recessive genetic disorders are frequent [21]. Due to the rarity of this disorder in the population and the complexity of the FV gene, little information is available about the molecular basis underlying FV deficiency. This study aimed to investigate the molecular and clinical characterization of 7 Iranian patients with severe FV deficiency.
Materials and Methods
Study population
Seven unrelated FV-deficient subjects from different regions of Iran who had been recognized at the Iranian Comprehensive Hemophilia Care Center (ICHCC) were enrolled. All patients signed informed consent, and the study was approved by the ethical committee of the Iran University of Medical Sciences, Tehran. All cases completed a comprehensive questionnaire containing demographic information, bleeding symptoms, and age of the first presentation, FV: C activity, received treatment, and familial history of bleeding.
Coagulation studies
The patients had previously been diagnosed as FV-deficient patients based on reduced FV coagulation activity test (FV: C) and routine coagulation tests at the ICHCC. The subject’s peripheral venous blood samples were collected into tubes in 1:10 volume of 3.8% sodium citrate. Plasma was obtained by centrifugation at 2000 g for 10 min and aliquots and
was stored at -80 °C until tested. FV antigen
in plasma was assayed with an enzyme-immunoassay kit AssayMax™. In addition,
for DNA isolation, blood samples in Ethylenediaminetetraacetic acid tubes were collected.
Amplification of F5 gene
High molecular weight DNA was extracted from whole blood using the extraction kit (Favorgen, Biotech, Taiwan). Primers were designed to amplify the entire F5 coding region, exon-intron boundaries, 5’ and 3’ untranslated regions (UTRs) based on the reported genomic DNA sequence of F5 (GenBank accession number Z99572). Because of the large size of exon 13, this exon was divided into 7 fragments and primer pairs were designed for each fragment. The primer sequences are available on request. Polymerase chain reaction amplifications were performed in a total volume of 25 µl using Taq DNA polymerase 2X mastermix (RED-Amplicon).
DNA sequence analysis
DNA sequence analysis was performed with chromas software (http://technelysium.com. au/wp/chromas/). According to Jenny et al. report, nucleotides and amino acids were numbered [16]. The already known mutations were mentioned by referring to the study which introduced them for the first time. For the novel mutations, molecular modeling was performed to simulate the molecular behavior of the mutant FV protein.
Results
Baseline information
Clinical data and laboratory measurements are summarized in Table 1. Patients generally had a moderate bleeding phenotype. Epistaxis was the most common sign in the probands.
Laboratory measurement
All the patients had FV activity <1% except for patient number 3, who had FV activity <2% (normal range: 82–118%). FV antigenicity of all subjects, excluding patients 3 and 4, whose plasma samples were not available, were measured, and the results are presented in Table 1(normal range: 90–110%).
Table 1. Phenotype characteristics of 7 Iranian patients with factor V deficiency.
Number
of cases |
Age/sex |
Age of first presentation |
Parental consanguinity |
Familial history |
FV:C |
FV:Ag |
Treatment |
Symptoms |
| 1 |
39/M |
1.5 |
+ |
+ |
<1% |
<5% |
FFP |
Severe epistaxis and gum bleeding
Severe soft tissue bleeding
Easy bruising
Bleeding after surgeries
Hemarthrosis |
| 2 |
33/F |
5 |
+ |
- |
<1% |
<5% |
FFP |
Epistaxis and gum bleeding
Menorrhagia
Easy bruising |
| 3 |
31/F |
28 |
+ |
- |
<2% |
NA |
FFP |
Recurrent miscarriages
Soft tissue bleeding |
| 4 |
41/M |
35 |
+ |
+ |
<1% |
NA |
FFP |
Gum bleeding
Epistaxis
Hematuria
GI bleeding
Severe bleeding after dental
Extraction |
| 5 |
45/M |
4 |
+ |
- |
<1% |
6% |
FFP, cryoprecipitate |
Epistaxis
Easy bruising |
| 6 |
50/F |
45 |
+ |
- |
<1% |
7% |
FFP, packed RBC |
Severe postmenopausal bleeding
Soft tissue bleeding
Epistaxis
Severe bleeding after dental
Extraction
Hemarthrosis
GI bleeding |
| 7 |
40/M |
27 |
+ |
+ |
<1% |
<5% |
FFP, cryoprecipitate, packed RBC |
Gum bleeding
Mucosal bleeding
GI bleeding |
M = Male; F = Female; GI = Gastrointestinal; FV: C = FV activity; FV: Ag = FV antigen; FFP = Fresh frozen plasma; RBC= Red blood cells; NA = Not available
V Mutations in patients with FV deficiency
Sequencing analysis of our patients’ PCR products revealed seven different mutations besides ten different single nucleotide polymorphisms (SNPs) (Table 2). Two of the mutations were splicing site mutations, one deletion, and the others were missense. Out of the identified mutations, three mutations were previously reported mutations (in patients 1, 2, and 3), including IVS 19+3 A>T [29], 4014-4017 del TCAG [30], and p.P419R [31, 32].
Table 2. Molecular characterization of 7 Iranian patients with FV deficiency
| Patient number |
Exon |
Nucleotide mutation |
Protein mutation |
Type of mutation |
Domain |
Genotype |
Reference |
SNPs |
| 1 |
- |
IVS 19+3 A>T |
1878-1901 del |
Splicing site mutation |
C1 |
Homozygous |
[29] |
C4185T |
| 2 |
13 |
4014-4017 del TCAG |
novel stop codon at position 1303 |
deletion |
B |
Homozygous |
[30] |
- |
| 3 |
9 |
c.1430 C>G |
p.P419R |
Missense |
A3 |
Homozygous |
[31] |
A2663G,A2684G, A2863G, A5380G, T3894C [33], C4038T (34] and C3943A |
| 4 |
- |
IVS9-1 G>C |
- |
Splicing site mutation |
A2 |
Homozygous |
This study |
C3943A |
| 5 |
10 |
c.1606 T>G |
p.Y478D |
Missense |
A2 |
Homozygous |
This study |
C4185T |
| 6 |
18 |
c.5705 T>C |
p.L1844P |
Missense |
A3 |
Homozygous |
This study |
A2663G, A2684G, A2863G, C4300T, A5380G, T3894C
C4038T, G409C |
| 7 |
13 |
c. 4841 T>C |
p.I1556T |
Missense |
A3 |
Homozygous |
This study |
- |
Four novel mutations were observed in the other four patients and were identified as IVS9-1 G>C, Y478D, L1844P, and I1556T. All mutations were named based on the official recommendations of the Human Genome Variation Society (HGVS, http://www.hgvs.org/mutnomen/ recs-DNA.html).
Molecular modeling of novel mutations
Four novel mutations were detected in 4 studied cases, including IVS9-1 G>C, Y478D, L1844P, and I1556T. Molecular modeling was done for Y478D, L1844P, and I1556T based on the FV PBD file downloaded from http://www.rcsb.org/. To compare structural changes in both normal and mutant states during simulation, Root mean square deviation (RMSD), Root Mean Square Fluctuation (RMSF), and Hydrogen bond (H-Bond) parameters were analyzed.
RMSD and RMSF
RMSD represents the number of changes in a structure relative to the reference structure. The reference structure is the zero-time structure of the simulation, and other structures were compared with this structure over time. In this section, the displacement of Cα atoms as a representative of the protein over a time of 50 nanoseconds compared to the first structure in all four simulations (normal and three mutants) was analyzed (Figure 1).
After the time 10,000 picoseconds (ps), the behavior of the graph was relatively different in all four simulations. According to Figure 1, in natural protein after this time, the amount of RMSD showed a slight decrease. Furthermore, in a time of about 11,000 ps, it reached 0.44 nm, and in the continuation of the simulation, the value of RMSD showed a relative increase again. In the Y478D mutation, the upward trend of the RMSD diagram slowed down after the time of 10,000 ps and at the time of 40,000 ps from the simulation, it reached 0.8 nm and showed a slight fluctuation around this value until the end of the simulation.
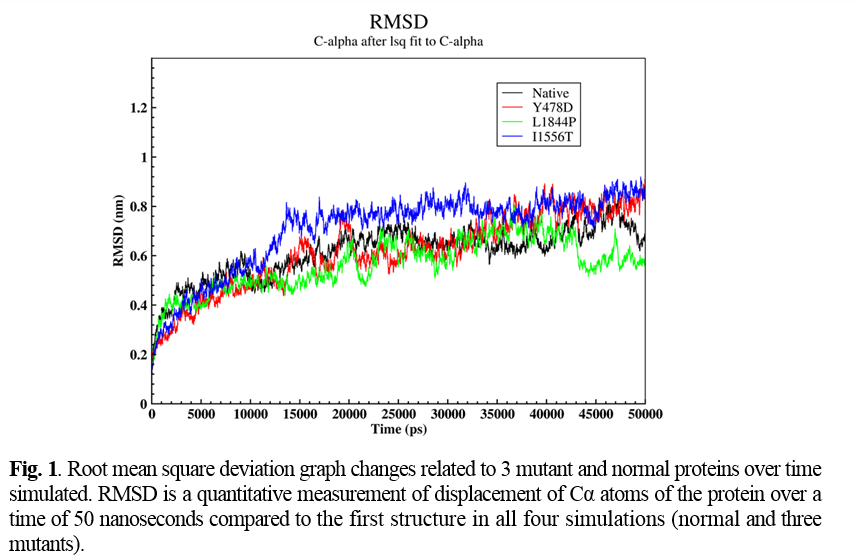
In the L1844P mutation, after 10,000 ps, the RMSD of the protein continued to rise, reaching about 0.64 nm at 20,000 ps. Then there was a sudden decrease in the amount of RMSD of the protein so that in about 22,000 ps, the amount of RMSD of the protein was equal to 0.44 nm, and then the amount of RMSD again showed a significant increase, and again it reached 0.64 nanometers in 23,000 ps. After 42,000 ps, the RMSD decreased to 0.48 nanometers at 43,000 pc and remained constant until the end of the simulation.In the I1556T mutant protein simulation, the RMSD value of the protein showed a significant increase after 10,000 ps. At 13,000 ps, it reached 0.78 nm, and after this time, the upward trend slowed down so that at 45,000 ps, the RMSD value of the protein reached about 0.8 nm and remained constant at this value until the end of the simulation.
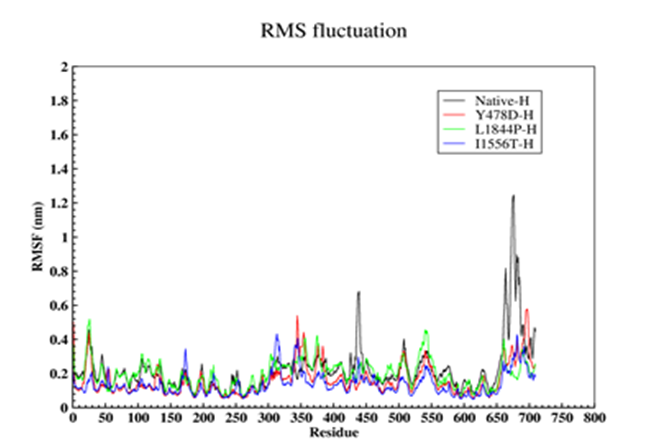
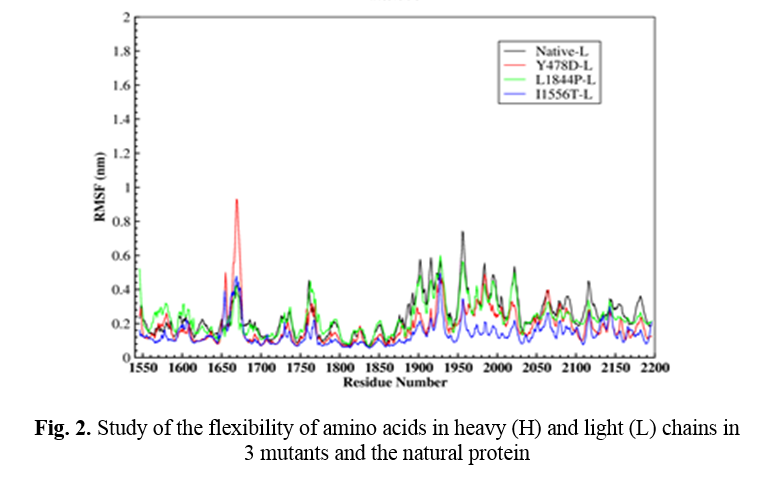
In order to further study the stability of all four proteins, the fluctuation value of each amino acid was evaluated. The dynamic behavior of alpha Cα in the protein structure contained sufficient information to study important motions in proteins and reflected the general motions of the structure. The RSMF of Cα atoms was considered to study the motion and structural flexibility. This section performed a structural flexibility comparison between the natural protein and the three mutant proteins (Figure 2). As shown in Figure 2, in the heavy chain in the two regions of 440-430 and 690-660, the flexibility of the three mutated proteins was greatly reduced. In the Y478D mutation, the flexibility in most areas was less than the normal protein and only in the range of 344-355; the flexibility of this protein was more than the natural protein. In the L1844P mutant protein, the degree of flexibility in most areas was similar to the natural protein, and only in two regions 490-480 and 540-530 the amount of flexibility of mutant protein was more than the natural protein.
In the I1556T mutation, the degree of flexibility in most areas was less than the natural protein and the other two mutants, and only in the two regions of 176-170 and 310-320, the degree of flexibility of this protein was higher than the other proteins. In the light chain, the amount of 3 mutants’ protein flexibility was less than the natural protein, with a few exceptions.
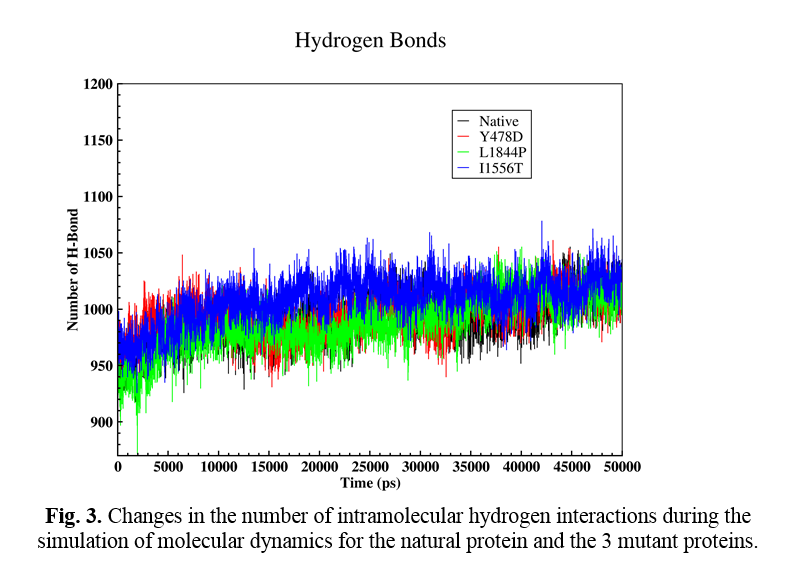
Hydrogen interactions
The effect of mutation on changing the number of intramolecular hydrogen interactions was also analyzed. Hydrogen interactions are one of the most important factors in protein stabilization. Figure 3 shows the changes in the number of H-bonds during the molecular dynamic simulation time (Fig. 4). The number of H-bonds in the natural protein and mutations Y478D, L1844P, and I1556T are estimated to be 991, 995, 988, and 1009, respectively. According to the obtained numbers, it may be said that in the L1844P mutation, the number of interactions is three bonds less than the normal protein, and in the Y478D mutation, four increased H-bonds are observed. However, in the I1556T mutation, the number of H-bonds increased by 18.
Solvent accessible surface area change (SASA)
The protein SASA factor indicates the number of amino acids exposed to the solvent. In the Y478D mutation, the total surface value at the end of 10 nanoseconds (50,000-40000 pcm) shows a slight increase compared to the natural protein, and this increase is due to the increase in the hydrophilic surface of this protein compared to the natural protein. In the other two mutations, the total surface relative to the natural protein decreased, which mainly decreased due to the decreased hydrophobic levels.
Based on the results of the performed analyses, it may be concluded that in the two mutations Y478D and I1556T, the number of H-bonds compared to the natural protein shows an increase, which reduces the flexibility and increases the stability of the protein. The results also show that in L1844P and I1556T, the solvent-accessible surface area (both hydrophilic and hydrophobic) of the mutant proteins show a significant decrease compared to the natural protein. On the other hand, the hydrophilic surface of the protein with Y478D mutation is increased compared to the natural protein.
Discussion
Severe coagulation FV deficiency is a rare inherited bleeding condition with relatively mild to moderate bleeding tendency. However, cases with life-threatening bleeding such as intracranial hemorrhage, gastrointestinal bleeding, recurrent miscarriage, and hemarthrosis have been reported [22, 25]. The Iranian study performed by Lak et al. [21] divided their 35 FV-deficient patients into three categories, including severe with FV: C<1%, moderate with FV: C 2-5%, and mild (FV: C 6-10%).
Patients in the severe group have experienced epistaxis (62.5%), menorrhagia (75%), haemarthrosis (31.25%), muscle hematoma (43.7%), Central nervous system bleeding (12.5%), post-operative post-partum bleeding (43.7%), oral cavity bleeding (56.25%) and haematuria (6.25%).
In this study, we investigated seven patients with severe quantitative FV deficiency. All the subjects had less than 1% FV activities except for patient number 3, who had <2% activity. FV antigenicity for the subjects was measured, excluding patients 3 and 4 whose plasma samples were not available. All studied cases had reduced FV activity and antigenicity together; their deficiencies were type I factor V deficiency. All patients’ parents were related, and consanguineous marriage is an important factor in the incidence of FV deficiency like other congenital bleeding disorders. All seven patients were symptomatic and suffered from a variety of mild to severe bleeding symptoms. Patients number 1, 6, and 7 had experienced several severe bleeding symptoms in their lives. All seven patients had experienced frequent epistaxis, especially during childhood. It is consistent with Lak et al., who reported that 57% of their patients had experienced frequent epistaxis. Severe gastrointestinal bleedings occurred in 2 patients, and two patients had damaged joints because of hemarthrosis. Oral cavity bleeding and hematoma were frequent in our patients. One patient has had two recurrent miscarriages so far because of severe genital bleeding. Although our patients suffered from type I coagulation FV deficiency, significant differences were seen in their clinical symptoms, and some of them had mild phenotypes (patients 2 and 5) contrary to what was expected due to the low levels of FV [7, 18]. In addition, it is interesting that mice with undetectable FV antigen levels die in the uterine or immediately after birth because of massive hemorrhage, but in humans, the same insignificant FV antigen guarantees the survival of patients [35].
In 1994 the first missense mutation associated with FV deficiency was reported and named FV New Brunswick. This mutation was caused by the replacement of Ala 221Val in the A1 domain of FV, and in the plasma of these individuals, the factor V antigen level was reduced to 39% of normal and its activity to 26% of normal [36]. From 1994 until now, more than 150 mutations in FV-deficient patients have been reported, and most of them are listed in some databases. About 50% of reported mutations are missense mutations, and this list is constantly growing. Most of them are in the A and C domains.
Despite the high prevalence of FV deficiency in Iran, not many studies have been performed on factor V deficiency. In the study conducted by Lak et al. on Iranian patients with congenital defects of coagulation factor V (with plasma levels between 1% and 10%) to assess clinical symptoms. It was shown that haemarthroses and muscle haematomas were less common, even in severely deficient patients. More severe symptoms such as gastrointestinal and central nervous system bleeding were also rare. The severity of bleeding symptoms was only partially related to the degree of factor V deficiency in plasma [21]. A study by Asselta et al. [29] aimed at determining the responsible mutation in a 19-year-old Iranian patient with severe FV deficiency showed IVS19 + 3A → T mutation in the patient (which was detected in one of our patients too). Recently in 2020, the most extensive case series of patients with FV deficiency was done, and 9 Iranian FV-deficient patients were identified [32].
The present study, overall, seven mutations, including three already reported mutations (IVS 19+3 A>T, 4014-4017 del TCAG, and p.P419R) and four novel point mutations (IVS9-1 G>C, Y478D, L1844P, and I1556T) were identified in our patients in the homozygous state. In addition some previously described SNPs such as A2663G, A2684G, A2863G, A5380G, C4185T, C4300T [7], T3894C [33], C4038T [34], C3943A [37] and G409C [38] were also seen in our cases.
IVS 19+3 A>T was reported by Asselta et al. in an Iranian patient. They investigated the effect of this mutation on both protein and mRNA levels. They demonstrated that this substitution in the splicing site of exon 19 resulted in skipping entire exon 19 with a length of 72-bp; the resulting FV protein lacked the initial 24 amino acids from 1878 to 1901 C1 domain. The mutant FV protein expressed by COS-1 cells has impaired secretion and stability. So, this omission resulted in a severe defect in protein secretion in this patient, and it is an apparent reason for decreased FV antigen level in plasma [29]. Homozygous 4bp deletion of a direct repeat of four nucleotides in exon 13 of F5 gene of patient two was introduced already by Guasch et al. This deletion causes an abnormal sequence of 23 amino acids residue 1302 compared to normal FV and then a novel stop codon at position 1303. The result of this aberrant stop codon is a truncated FV protein with 1302 amino acid residue and without part of the B domain and the complete light chain [30]. Like Guasch et al.’s patient, our patient’s bleeding phenotype is mild despite the drastic defect in their FV protein. P419R in exon 19 was another identified mutation in this study. It was reported by Borhany et al. in a Pakistani woman. They predicted that P419R conversion changed the size and hydrophobicity of FV and resulted in FV damage. This mutation seems frequent in Pakistan as it was found in two unrelated probands in their study [31]. Our patient is from Sistan province, a region in the East of Iran, near Pakistan. Recently P419R was identified in another severe FV-deficient patient in a compound heterozygote state and introduced as a deleterious mutation [32].
A novel homozygous G>C single nucleotide point mutation (IVS9-1 G>C) at position -1 of one of our subject’s intron 8 of F5 was detected. This nucleotide substitution is likely to impair pre-mRNA splicing. Splicing mutations account for 10% of total F5 mutations, often associated with more severe forms of FV deficiency [39]. Splicing site mutations often result in a frameshift and relevant exon skipping due to premature stop codon. These truncating mutations condemn the transcribed mRNA to nonsense-mediated mRNA decay pathway (NMD) [29, 40]. To find out the exact impact of the F5 IVS9-1 G>C mutation on pre-mRNA splicing, an experimental investigation of the patient’s F5 cDNA analysis is required.
Y478D was another identified mutation in this study. RMS fluctuation analysis during molecular modeling shows that Y478D mutant protein generally has decreased flexibility compared to the natural protein in heavy and light chains. Y478D mutant protein has 4 H-bonds more than the natural protein, and it helps to excess stability of this protein compared to the wild type one. The hydrophilic surface value of Y478D mutant FV is more than the natural protein, leading to the increased total surface area.
L1844P was detected in one of our patients with a severe bleeding phenotype. According to the molecular dynamic simulation performed for this mutation, the solvent-accessible surface area (both hydrophilic and hydrophobic) showed a significant decrease compared to the natural protein. There is a not noticeable change in the H-bond and flexibility of the L1844P mutant protein and the natural one.
Patient number 7 showed I1556T mutation in his FV protein. Significant increase and reduction in RMSD and RMSF value (in both H and L-chains) respectively and increase in the number of H-bonds to 1009 (18 more bonds) in I1556T mutant protein compared to the natural protein confirm the decrease in flexibility and increase in the stability of the mutant protein. The total surface of I1556T mutant FV relative to the natural protein decreases in hydrophilic and hydrophobic surfaces.
According to previous studies, F5 mutations are specific to each family, and except for a few mutations such as Y1702C [41], frequent mutations are not seen in unrelated patients.
Further studies are needed to understand the exact association between these novel mutations and patients’ clinical symptoms.
We are identifying the responsible mutation in patients with FV deficiency aid to determine the molecular basis of this bleeding disorder and obtain more insight into the clinical heterogeneity of severe coagulation FV deficiency. In addition, molecular diagnosis in FV-deficient patients is essential to genetic counseling and prenatal diagnosis.
Conclusion
Identifying the causative mutation in patients with FV deficiency helps to determine the molecular basis of this bleeding disorder and gain more insight into explaining the variable clinical manifestations of patients with FV deficiency.
Conflict of interest
The authors have no conflict of interest to declare.
Acknowledgments
This study was supported by grant No. 13579 by the Iran University of Medical Sciences.
References