
Biotechnology Research Center, Yazd Reproductive Sciences Institute, Shahid Sadoughi University of Medical Sciences and Health Services, Yazd, Iran
Full-Text [PDF 545 kb]
(57 Downloads)
|
Abstract (HTML) (106 Views)
Full-Text: (80 Views)
Recurrent pregnancy loss (RPL) is commonly defined as three or more consecutive unsuccessful pregnancies before 22 weeks of gestation, affecting approximately 2-3% of women who intend to become pregnant [1]. While severe fetal malformations, caused mainly by chromosomal abnormalities, have been established as the proven factor in RPL, in many cases, no clear cause for RPL can be identified. The other known risk factors for such pathological conditions are infections, Antiphospholipid Antibody Syndrome (APS), anatomical problems, and immunological and genetic factors [2]. According to the literature, 40% to 50% of RPL cases are classified as idiopathic or unexplained RPL with unknown etiology. Various factors have been suggested for these cases, which can be divided into two general groups: environmental factors, such as ovarian aging, unknown alloimmune factors related to the environment, and genetic factors affecting immune system function [3].
In RPL studies, the genetic material from two generations, the mother/father and the offspring, is usually analyzed. This involves examining the expression of genes and proteins in different compartments, namely the mother, placenta, and fetus. Gene/protein expression Aberrations can impact maternal and fetal organisms [4]. According to the Euro-Team Early Pregnancy protocol for RPL, it was suggested to perform a cytogenetic investigation of the products of conception (POC) material if a couple experiences two spontaneous miscarriages and both partners have normal karyotypes. This helps in providing prognostic information and to assess the efficacy of treatment. However, routine POC genetic testing has not yet been widely adopted due to the challenges in obtaining biological material suitable for in vitro culture, a prerequisite for karyotyping [5].
As a result, in the last two decades, various targeted molecular assays have been developed for the detection of aneuploidy from uncultured material, such as fluorescent in situ hybridization (FISH), multiplex ligation-dependent probe amplification (MLPA), and quantitative fluorescent polymerase chain reaction (QF-PCR). Furthermore, with cutting-edge technologies such as array comparative genomic hybridization (aCGH) and single nucleotide polymorphisms (SNP) microarrays, it is now possible to conduct high-resolution profiling of microdeletions/duplications on a genome-wide scale. In addition, next-generation sequencing (NGS) technology offers single base pair resolution and the most sensitive detection of mosaicism among all available methods [6].
Genetic factors involved in RPL
Thus far, various genetic factors have been identified as underlying causes of RPL. Most studies have categorized genetic factors according to their maternal or paternal source. Nonetheless, despite some factors being passed explicitly from one parent, many can be inherited parallelly from both parents, leading to RPL. Furthermore, the lack of RPL in the siblings of both couples experiencing this issue suggests that the combination of genetic profiles from two families comprising the fetal genomic background may be responsible for this pathological state. Accordingly, the genetic factors leading to RPL have been reviewed in this study in 4 groups. Figure 1 illustrates these genetic factors and their origins.
Paralleled maternal/paternal genetic factors
Many studies have tried to identify the genetic factors that cause RPL and have categorized them based on whether they come from the mother or father. However, there is often no distinction between specific maternal or paternal factors and genetic factors inherited in parallel from both parents. These jointly inherited genetic factors could come from two sources: 1) genetic factors derived from the maternal or paternal genome, including chromosomal abnormalities, mutations that affect cellular processes, and detrimental alleles of common genetic variations, such as those found in thrombophilic and immunoregulatory genes; 2) genetic factors that arise during the gametogenesis process in the maternal or paternal germ layer, including chromosomal abnormalities and mutations that affect cellular processes.
Maternally/ Paternally inheritable genetic factors
Chromosomal abnormalities
Chromosomal abnormalities are a significant genetic factor that contributes to RPL. These abnormalities can be inherited from both mother and father [7] and affect the process of implantation, maintaining the embryo, and the development of the fetus. However, the impact of maternally and paternally inherited factors can vary.
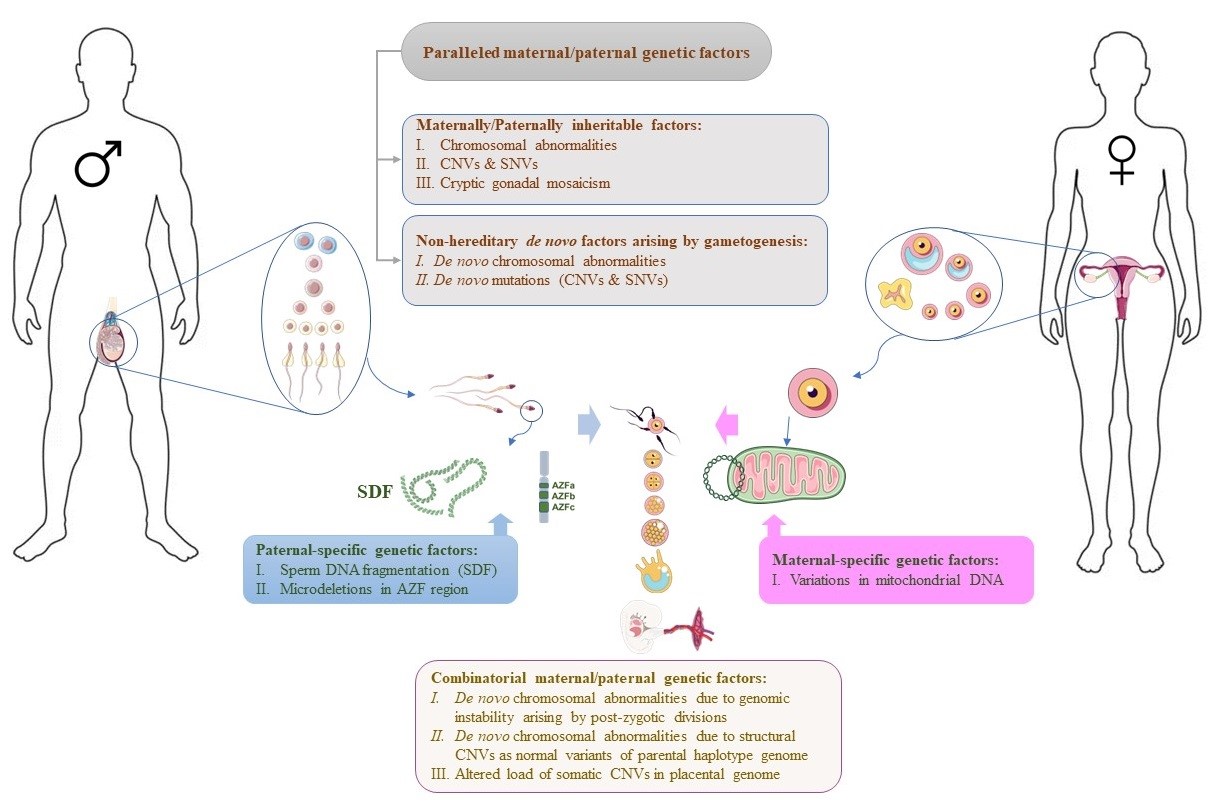
Fig. 1. The schematic image illustrates the genetic factors involved in RPL and their genomic origins. The Figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
While maternal chromosomal abnormalities can affect the embryo from the beginning of fertilization and during oocyte maturation, the paternal genome activation and subsequent zygotic genome activation (ZGA) occur at the 4-8 cell stage in human embryos. Therefore, genetic abnormalities in sperm (variants of paternal origin) may not affect pre-ZGA embryonic development but may relate to defects in later embryonic development or abnormal intrauterine fetal development [8]. The rate of gross chromosomal abnormalities in either or both parents within the RPL population is approximately 2-4%, including asymptomatic individuals with balanced translocations or inversions [9, 10]. However, according to recent studies by Ahangari et al. [11] and Pal et al. [12], couples who suffer from RPL are more likely to carry chromosomal abnormalities, with a rate of 3.74% to 9.88%. The most common chromosomal abnormality in RPL couples is balanced translocation, which accounts for 38% to 47.05% of cases. Inversions account for 29.41% to 34.70% of cases, followed by numerical abnormalities (11.76% to 16.50%) and Robertsonian translocations (10.70% to 11.76%), as reported by different groups [11-13]. Interestingly, female carriers of chromosomal aberrations tend to be more frequent than male carriers among RPL couples [11, 14].
Submicroscopic genomic rearrangements
Diagnostic techniques based on microarray technology have revealed new molecular insights from the analysis of submicroscopic chromosomal variations. These variations, known as DNA copy number variants (CNVs), include deletions, duplications, and complex rearrangements of chromosomal regions. They can range in size from a few hundred base pairs (bp) to several megabytes (Mb) [5]. Research on CNVs in RPL families has provided new molecular and clinical insights in recent years. For more than ten years, the association of some CNVs, especially pericentromeric abnormalities, with RPL has been identified [15, 16]. Several studies have reported a positive correlation between the overall burden of CNVs and the risk of miscarriage [17, 18]. Furthermore, recent studies have shown an increased prevalence of large pericentromeric and sub-telomeric CNVs in the genomes of female and male partners of RPL couples [17, 19]. However, the impact of these structural genomic variants on pregnancy-related disorders may vary based on their size, type, and chromosomal location, as well as the number of genes involved and whether they affect the early pregnancy period [5].
The analysis of the underlying genes of CNVs identified in the genome of parents with RPL shows that most of these variants are related to the function of the immune system, such as allograft rejection, complement cascade, antigen binding, and antigen presentation (Table 1) [17, 18, 20]. A few recurrent CNVs have been identified as associated with RPL risk, highlighting novel genes and mechanisms possibly implicated in RPL development. For example, a 60 kb duplication encompassing the GOLPH3 and PDZD2 genes (with a regulatory role in Golgi trafficking) has been identified with a 5-fold higher prevalence among RPL females than fertile women [18].
Single nucleotide variants
In recent years, NGS methods, particularly whole exome sequencing (WES), have helped advance our understanding of RPL. These methods have identified a range of gene variants that underlie the disease. While most studies have focused on identifying RPL-associated mutations in female partners, several studies have also examined the paternal genome, revealing the role of paternal heritable variants in the pathogenesis of RPL [8]. According to Cao et al.’s report, until 2022, fourteen studies using exome sequencing have been conducted to identify potential genes and mutations associated with RPL in affected couples. The findings of these studies have linked a total of 66 maternal and 12 paternal genes with RPL. However, only two genes - KHDC3L and CCNB3 - have been repeatedly reported in more than one study, highlighting the complex nature of RPL as a multifactorial disease [8]. The identified genes are involved in different functional categories of biological processes, including coagulation and angiogenesis, extracellular matrix synthesis and degradation, regulation of immune responses, and cell divisions (meiosis and mitosis) and related processes, such as DNA replication, DNA repair, and chromatin remodeling. While findings from these studies provide new insights into the pathogenesis of RPL, functional studies are needed to determine the contribution of these genetic variants in the disease and develop novel strategies for RPL management [5, 8].
Although genome-wide studies have revealed unknown single nucleotide variants (SNVs) in RPL families, many studies have focused on the genotyping of targeted SNVs. These studies have evaluated the known genetic variations that affect biological pathways potentially involved in RPL, such as hormonal regulation, detoxification systems, thrombosis and cardiovascular system, and, more frequently, the immune response and inflammation [4]. However, the association of SNVs with RPL has been highly variable among studies, possibly due to the clinical criteria assigned for RPL definition and genetic models used in such studies [5].
Three extensive systematic reviews have examined more than 100 genes and approximately 500 SNVs, mainly conducted on female partners of RPL families [4, 21, 22]. Interestingly, all three studies reached a relatively consistent conclusion that most nearly 500 common SNVs analyzed had no significant correlation with RPL. However, some variants were modestly correlated with the increased risk of developing RPL, including those related to thrombosis and metabolism, as well as the SNVs led to over-active immunological responses, such as those investigated in the genes interferon‐gamma (IFN-γ), interleukin (IL)-10, KIR2DS2, KIR2DS3, KIR2DS4, MBL, and tumour necrosis factor (TNF)-α. The majority of these variants, about two-thirds, have only been identified in one study. These observations may be attributed to incidental associations, the cumulative impact of other unspecified genetic factors, and environmental conditions influencing the onset of the disease [5]. Therefore, further studies are warranted to uncover the role of these factors.
The known SNVs in the two significant pathways of thrombosis and immune regulation, which have a profound impact on the process of abortion, will be further investigated in the following sections.
Thrombophilic factors
Several studies have suggested that thrombophilia, a condition where the blood has an increased tendency to clot, can increase the risk of RPL in women by impairing normal placental vascular function. It’s important to note that the thrombosis or occlusion of placental vessels can decrease placental perfusion, which can lead to adverse pregnancy outcomes, including RPL. It’s widely accepted that thrombophilia is a common risk factor for RPL and can be observed in about 40% to 50% of RPL cases [23]. Inherited variants in some thrombophilic factors have been extensively investigated for their associations with RPL, including factor V (FV), prothrombin, and homocysteine metabolism-associated enzymes, namely methylenetetrahydrofolate reductase (MTHFR), methionine synthase (MTR), and methionine synthase reductase (MTRR) [24]. It is also has been found that both paternal and maternal mutations of thrombophilic genes, including Factor V Leiden (FVL), Factor V receptor 2 (FVR2), angiotensin I converting enzyme (ACE), and Apolipoprotein E2 (ApoE2), are related to RPL [25].
ANXA5 is an anticoagulant protein found at high levels in the human placenta. The M2 haplotype of the ANXA5 gene has been identified as a hereditary thrombophilic genetic factor linked to RPL [26]. Research has shown that carriers of the M2 haplotype in the ANXA5 gene promoter region have more than two times higher RPL risk than the general population [27]. Besides RPL, studies have also proved the association of this haplotype to various obstetric pathologies, including gestational hypertension, preeclampsia, fetal growth restriction, preterm birth, and antiphospholipid syndrome [28]. Remarkably, the paternal ANXA5 M2 haplotype has been found to have a similar effect on the risk of RPL as the maternal mutation [29]. New findings indicate that embryos carrying the M2/ANXA5 haplotype inherited from both parents are susceptible to recurrent implantation failure (RIF) [28].
Immunoregulatory factors
During pregnancy, the fetus expresses the antigens of paternal origin, making it “foreign” to the mother’s immune system. This means immune tolerance between the mother and fetus is crucial for a healthy pregnancy. Most studies in this area have focused on genetic variants of maternal origin, as the mother’s immune system is responsible for “rejecting” the semi-allograft fetus [8]. However, recent research has focused on the immunological role of fetal cells (originating from both the father and mother) affecting the immunoregulatory process. In particular, the human leukocyte antigens (HLA)-G has a restricted expression in the EVT and functions at the maternal-fetal interface. HLA-G, a non-classical MHC Class I antigen, is coded by a gene that generates several isoforms. These isoforms are essential in sustaining a healthy maternal-fetal interface throughout pregnancy [30]. Studies on the null allele of HLA-G isoforms proved that insufficient levels of the HLA-G proteins may negatively impact the outcome of a pregnancy [31]. In light of previous findings indicating minimal variability in its coding regions, attention has shifted to exploring the non-coding regions of the gene (Table 1). Surprisingly, conflicting reports have emerged on the correlation between maternal HLA-G gene variants and RPL in these investigations [8]. The role of the paternal genome and inherited alleles from the father appears to have been undervalued in this scenario.
Studies have shown that numerous polymorphisms in various cytokine genes, including in pro-inflammatory (IL-1β, IL-2, IL-17, IL-18, TNF-α, and IL-33) and anti-inflammatory genes (IL-10, TGF-β1 and IL-6), are associated with the susceptibility to RPL [8]. Moreover, several variants in genes involved in the development and function of Regulatory T cells (Tregs) cells, notably the transcription factor FOXP3, are also significantly associated with RPL susceptibility [32, 33]. Activation of naïve T cells leads to the induction of effector T cells, which play a crucial role in regulating T cell responses and immune tolerance. Key negative regulators of this pathway, including cytotoxic T lymphocyte-associated protein 4 (CTLA-4), programmed cell death 1 (PD1), and PD-ligand 1 (PDL1), inhibit proliferation and cytokine production in T cells. Multiple variants in these genes have been associated with RPL [34-36]. Furthermore, earlier immunological evidence has shown that the complement system plays a vital role in pregnancy. Recent findings also suggest that specific genes in complement pathways, such as complement factors D (CFD) and H (CFH), are positively correlated with an increased risk of RPL [37]. Some variants related to the genes of pro-inflammatory and anti-inflammatory factors, key regulators in the induction of Treg cells and effector T cells, and complement factors are summarized in Table 1.
Table 1. The maternally/paternally inheritable variants involving RPL-related immunoregulatory processes.
|
Categories |
Genes |
Variants position |
Mutation type |
Ref. |
| SNV |
Immunoregulatory function in lymphocytes |
FKBP4 |
c.47C>A (p. A16E) |
missense |
[38] |
| Activation of pro-inflammatory caspases by inflammasomes |
NLRP7 |
c.1857_1858delAC (p.Lys619Asnfs)
rs2069070395 |
frameshift |
[39] |
| Regulation of innate immune system |
NLRP10 |
c.1015G>T (p.D339Y)
rs144864261 |
missense |
[40] |
| TLR3 |
c.2384C>T(p.Ala795Val)
rs373118024 |
missense |
[41] |
| Activation of cytokine promoters such as the IL-13 and TNF-α |
NFAM1 |
c.522C>A (p. R189Ter)
rs34963472 |
stop gained |
[42] |
c.498T>G/C (p.C181G/ R)
rs5996153 |
missense |
| Cytokine receptor on Macrophages |
CSF1R |
c.2498C>T (p.T833M)
rs780804532 |
missense |
[40] |
| Immunoglobulin superfamily |
PSG9 |
g.43772302C>T
rs3746297 |
splice-acceptor |
[43] |
| HLA class I |
HLA-G |
−725 C/G/T |
5′ UTR |
[31] |
−1573 T/C
−1746 C/A |
[44] |
| 14 bp deletion/insertion |
3′ UTR |
[45] |
| Pro-inflammatory cytokines |
IL-1β |
g.4490T>C |
Upstream Variant |
[46] |
| IL-2 |
g.4671T>G |
Upstream Variant |
| IL-17 |
g.4849G>C |
Upstream Variant |
[47] |
| IL-18 |
137 G/C |
|
[48] |
| IL-33 |
c.-11-12267G>A |
Intron Variant |
[49] |
| Anti-inflammatory cytokines |
IL-10 |
g.6195A>G |
Intron Variant |
[50, 51] |
| −819 C/T |
|
| IL-6 |
g.4481G>C |
Upstream Variant |
| IL-22RA2 |
c.610 C>T |
|
| TGF-β1 |
c.74G>C
p. Arg25Pro |
Missense Variant |
[52] |
| TNF-α |
g.4127C>G |
Upstream Variant |
[53] |
| g.4682G>A |
Upstream Variant |
| Treg cells |
FOXP3 |
c.1540-18T>A |
Intron Variant |
[32] |
| c.-23+2877C>T |
Intron Variant |
| c.-22-902A>T |
Intron Variant |
| CD8⁺ CTLs |
CTLA-4 |
c.49A>T
p. Thr17Ser |
missense |
[54] |
| Immune inhibitory receptor/ligand on lymphocytes |
PD1 |
g.4463G>A |
5′ UTR |
[36] |
| PDL1 |
Ivs2 +6371 G>A |
Non-coding |
[55] |
| Complement system |
CR1 |
c.4501A>G (p.T1501A)
|
missense |
[41] |
| CFH |
c.2325G>T (p.E775D)
rs1065489 |
missense |
[37] |
c.1204C>T (p.H402Y)
rs1061170 |
missense |
| CFD |
c.744C>G (p.I248M)
rs2230216 |
missense |
| CNV |
8p23.1,
10q11.21-q11.22
15q11.2 |
macro-alteration |
[17] |
allograft rejection signaling
IL-4 signaling
autoimmune thyroid disease signaling |
GSTT1 |
22q11.23 |
microdeletions/amplifications |
| CTDSPL |
3p22.2 |
| HLA |
6p21.32 |
| MSR1 |
8p22 |
| Immunoregulatory pathways essential for immune tolerance at fetomaternal interface |
PDZD2
GOLPH3 |
multicopy duplication (61.6 kb) at 5p13.3 |
|
[18] |
SNV= Single nucleotide variant
CNV= Copy number variants
FKBP4= FKBP prolyl isomerase 4
NLRP7= NLR family pyrin domain containing 7
NLRP10= NLR family pyrin domain containing 10
TLR3= Toll like receptor 3
NFAM1= NFAT activating protein with ITAM motif 1
CSF1R = Colony stimulating factor 1 receptor
PSG9= Pregnancy specific beta-1-glycoprotein 9
HLA-G = Major histocompatibility complex, class I, G
IL-1β = Interleukin-1 beta
IL-2= Interleukin-2
IL-17= Interleukin-17
IL-18= Interleukin-18
IL-33= Interleukin-33
IL-10= Interleukin-10
IL-6= Interleukin-6 |
IL-22RA2= Interleukin 22 receptor subunit alph
TGF-β1= Transforming growth factor beta 1
TNF-α = Tumor necrosis factor alpha
FOXP3= Forkhead box P3
CTLA-4= Cytotoxic T-lymphocyte associated protein 4
PD1= Programmed cell death 1
PDL1= Programmed death-ligand 1
CR1= Complement C3b/C4b receptor 1
CFH= Complement factor H
CFD =Complement factor D
GSTT1= Glutathione S-transferase theta 1
CTDSPL= CTD small phosphatase like
HLA= Human leukocyte antigen
MSR1= Macrophage scavenger receptor 1
PDZD2= PDZ domain containing 2
GOLPH3= Golgi phosphoprotein 3 |
|
|
|
|
|
|
|
Non-hereditary de novo genetic factors
Apart from inheriting identical portions of the genome from both parents, each developing embryo carries a few distinct genetic disorders, which arise during gametogenesis and postzygotic development. De novo-generated mutations and chromosomal abnormalities happen due to errors in DNA replication/repair mechanisms and the chromosome segregation process. The de novo mutations are one of the causes of genetic variation in the population, whereas de novo chromosomal abnormalities are the main cause of sporadic miscarriages. These errors result in variable disorders, from SNVs and indel mutations to CNVs and microsatellite instability, genomic rearrangements, and chromosomal aneuploidies [56]. The occurrence of de novo genetic disorders in the zygote can result in varying levels of mosaicism, determining their clinical outcome. The consequences of the disorders depend on the developmental stage and the type of cells involved. De novo mutations can occur at different developmental stages, ranging from parental germline to postzygotic, embryonic, fetal, and post-natal events. Mutational processes can also occur in various cells in a tissue, including differentiating cells, stem cells, and terminally differentiated somatic cells. These factors can determine the tissue-specific distribution and burden of de novo disorders that can influence the potential disease they cause. Indeed, the timing of these disorders can generate variation, from homozygosity to heterozygosity, by affecting mutation load and distribution throughout the body [57].
It is widely acknowledged that parental gametogenesis, comprising oogenesis and spermatogenesis, is the primary site where errors in genome replication/repair and chromosome segregation can result in various de novo genetic disorders in each subsequent generation. Although it is well-known that the incidence of these disorders increases with the couple’s age, the contribution of each parent to their occurrence is different. The male germline undergoes 23 mitoses every year. This means that, for instance, a spermatogonial stem cell in a 40-year-old man has undergone over 600 mitoses. In contrast, the number of mitotic divisions remains constant during oogenesis. However, genetic disorders have been found to accumulate in oocytes over time, possibly due to the failure of the DNA repair process. Therefore, the higher rate of mitotic divisions in the paternal germline suggests that the father has a more prominent role in the occurrence of de novo genetic disorders in the fetus than the mother [58].
De novo chromosomal abnormalities:
Numerous investigations focusing on aborted fetuses have conclusively demonstrated that major chromosomal aberrations are the prevailing reason for both sporadic miscarriages (combined prevalence from 13 studies, 45%) and RPL (six studies, 39%, 29%–50%) [59, 60]. In a recent study that examined the rate of fetal chromosomal aneuploidies in POC samples, these abnormalities were reported to account for about 60% of spontaneous sporadic pregnancy losses [61]. According to a recent survey of 1000 POC samples, autosomal trisomies are the most prevalent type of chromosomal abnormalities, comprising 59.7% of cases. Polyploidies and monosomies follow, accounting for 22% and 7.5% of cases, respectively. Structural chromosome rearrangement and multiple aneuploidies are less common, representing 7% and 3.8% of cases, respectively [62]. However, when compared to sporadic spontaneous pregnancy loss, women with RPL have a lower embryonic chromosomal aneuploidy rate [63]. Most studies examining chromosomal aneuploidy in POCs have not determined the inheritance status of these abnormalities from carrier parents, so these findings cannot be considered “de novo” rates of chromosomal abnormalities.
While idiopathic RPL does not encompass miscarriages resulting from chromosomal abnormalities, the origin of genetically unbalanced gametes due to meiosis errors remains uncertain in many cases. In some cases, unbalanced gametes are produced due to carrier parents, particularly carriers of balanced translocations or cryptic mosaicism in the gonads that lack the phenotype. In other cases, these disorders arise de novo during parental gametogenesis. Although various environmental and genetic factors may play a role in de novo disorders, parents’ aging is still known as the main reason for this phenomenon. Indeed, as parents age, the chances of chromosomal abnormalities in their gametes increase, thus increasing the risk of miscarriage [64].
It is widely recognized that errors during mitosis and meiosis are common among all women. However, oocyte chromosomal abnormalities are caused mainly by random errors during maternal meiosis I (MI) [65]. Studies have shown that women who experience RPL and are of advanced maternal age consistently exhibit aneuploidy [66]. This may be due to de novo genetic disorders resulting from the missegregation of chromosomes during oocyte maturation [65]. It has been estimated that nearly 50% of oocytes are aneuploid [67], and 30% of conceptions are lost before a missed menstruation [5].
In addition to oogenesis, de novo genetic disorders during spermatogenesis have been identified as the reason for chromosomally abnormal embryos and the leading cause of embryonic mortality and RPL [68]. Previous studies have reported that 40% of men who demonstrated normal sperm density and motility and experienced RPL had sperm aneuploidy. Moreover, a higher proportion of sex chromosome aneuploidy has been observed in sperm samples from men with abnormal sperm density and motility compared to men with normal sperm parameters (62% vs. 45%) [69]. Recent studies further support the role of sperm aneuploidy in unexplained RPL [70-72].
De novo mutations
De novo mutations in humans are genetic modifications that have arisen within one generation. While most of the fetal genome is inherited from parents, DNMs are responsible for introducing novel genetic diversity. DNMs can take different forms, such as SNVs, insertions, deletions, and CNVs, with varying consequences [64]. Some DNMs are harmless or even beneficial, contributing to genome evolution. However, with the advent of NGS-based technologies that allow for whole-genome scanning, numerous DNMs have been identified as a major cause of malfunctioning biological systems that lead to neurodevelopmental diseases, including intellectual disability, autism, and schizophrenia [58, 73].
One of the most significant findings about DNMs is that their number increases with parental age during conception. DNMs can arise in various biological niches. An individual’s first DNMs develop during their parents’ embryonic development, starting with creating primordial germ cells (PGCs) during gastrulation. In males, DNMs develop in spermatogonial cells that continuously produce sperm as they age. In females, aging of the oocytes is not accompanied by genome replication, but mutations can still occur. For some genetic diseases, the association with paternal age is stronger than with maternal age. It is believed that the cause of this association could be imprecise genome copying during the numerous cell divisions required for continuous sperm cell production in males [64].
Most NGS-based discoveries regarding DNMs have been made using parental genomes on blood samples or fetal genomes on POC samples. This makes it difficult to determine whether the DNMs originated in the germ layer of the parents or if they arose post-zygotically in embryonic cells.
Maternal-specific genetic factors
Due to the maternal inheritance of mitochondria in the fetus, mutations and variants of the mitochondrial genome are known as maternal-specific genetic factors. Studies have revealed that mitochondria play a crucial role in the differentiation and activation of immune cells, and changes in mitochondrial DNA (mtDNA) are the primary cause of developmental disorders due to their involvement in ATP production and apoptosis [74, 75]. However, little is known about the association between mtDNA changes and RPL. One study compared mitochondrial mutations in women with RPL to those of age-matched control women. The results indicated a significant increase in heteroplasmic mtDNA diversity in the RPL group (27%) compared to the control group (19%). However, none of the detected variations in this study were previously known to be pathogenic and, therefore, unlikely to be the cause of miscarriage [76]. Another study analyzed the complete mtDNA of 100 women with RPL and tissue from 12 aborted fetuses. The results showed a significantly increased number of variants in the RPL group, particularly in the NADH dehydrogenase (ND) genes, which encode the complex I mitochondrial enzyme. The presence of the T4216C (ND1 gene) variant in 9% of RPL women and several novel pathogenic mutations suggest the role of mtDNA alterations in RPL [77].
Recently, mitochondrial respiratory chain defects, especially due to genetic changes in the genes encoding complex I in mtDNA, are known to be the main cause of some immune-related diseases [74] and the basis of carcinogenesis [75]. Therefore, such genetic changes are also considered as possible causes of RPL. For example, in a direct sequencing study, mtDNA mutations and variants were examined in women with idiopathic RPL. The results identified seven variants in mitochondrial complex I genes, four of which were synonymous (A5153G, C10142T, A14179G, and C14263T). The remaining three were non-synonymous (T4216C, C12063T, and A12662G) changes. The case idiopathic RPL group had significantly higher proportions of these variants than those observed in the control group [78]. In conclusion, further research is needed to understand the effect and role of mitochondrial variations in the progression of RPL, which may vary among individuals and ethnic groups.
Paternal-specific genetic factors
Certain genetic factors that contribute to RPL are inherited from the father. One such factor is the integrity of sperm DNA, which plays a vital role in fertilization and embryo development. When sperm DNA is damaged, a condition known as sperm DNA fragmentation (SDF) can lead to lower fertilization rates, terminated implantation, and miscarriages. Fertilization using damaged sperm can cause increased DNA damage in the fetal genome, leading to defects in various stages of embryogenesis and fetal development [79]. Recent studies have found that SDF has diagnostic value and suggests the possibility of paternal genetic factors in idiopathic RPL [80].
Also, Y chromosome microdeletions have long been considered a potential paternal genetic factor in idiopathic RPL. However, while some studies have linked microdeletions in a region known as azoospermia factor or Deleted in azoospermia with spermatogenic failure and male infertility [81, 82], others have found no association between Y chromosome microdeletions and RPL [83, 84]. Further research is required to explore the role of Y chromosome microdeletions as a male factor in the pathogenesis of RPL.
Combinatorial maternal/paternal genetic factors
With the development of advanced genomic analysis tools, it has become clear that RPL should be considered a disorder caused by intrinsic factors determined by the genome of RPL partners and/or as a result of their combination as POC. Recent evidence collected from the examination of POC samples shows that about 30-50% of these samples in RPL couples have gross chromosomal abnormalities or other structural genomic rearrangements that can describe the cause of miscarriage. The findings of recent years have shown that only 10% of these genomic abnormalities are directly inherited from parents, and 90% of them are caused by de novo genomic rearrangements in POC samples. Although some of these abnormalities arise during gametogenesis (oogenesis or spermatogenesis), as discussed earlier, another fraction of de novo genetic disorders result from postzygotic errors initiated during early embryonic development [5]. De novo disorders during gametogenesis do not seem to have the exact cause as postzygotic cases. The increasing age of parents is the main reason for the increasing rate of de novo disorders during gametogenesis [64], while postzygotic cases result from combining maternal and paternal genomes at POC [5]. It should be noted that there is usually no distinction between de novo disorders during gametogenesis and postzygotic types in clinical research, so most review studies discuss them together. However, it is important to recognize these disorders’ different causes to understand and treat RPL better.
Extensive research has shown that a considerable portion of aborted fetuses in cases of RPL, regardless of genetic abnormalities, exhibit developmental defects that make it unlikely for them to survive beyond the first half of pregnancy, including severe disturbances of growth and morphogenesis (e.g., embryonic growth disorganization) [85]. It has also been found that the risk of congenital severe disabilities is higher for infants born in RPL families (4.2%) than in those without a history of previous abortion (2.5%) [86]. A large study recently conducted on nearly 250,000 infants demonstrated the association between maternal history of RPL and long-term neurological morbidity and developmental and movement disorders of the offspring [87]. This is also supported by similar findings in other long-term pediatric diseases, such as respiratory morbidity of the offspring, which is possibly explained by a common immunological etiology [88]. Recent studies have proposed the hypothesis of “unfavorable genomes” as a possible consequence of combined maternal and paternal genomes on POC, leading to genomic instability in embryonic cells and a decreased chance of viable and healthy offspring for RPL couples [89]. Based on this, it has been suggested that the genomes of POCs in an RPL family carry unfavorable genomic factors that predispose them to severe developmental disorders in pregnancy [19]. Although these are currently hypotheses that require extensive studies to be confirmed, the recent evidence from examining POC samples supports this hypothesis. For example, analysis of RPL placental villus transcriptomes has exposed clear deficiencies in the vital elements that maintain the nuclear and early cellular machinery during the fast-paced proliferation and differentiation of embryonic cells. These findings confirmed the genomic instability in embryonic cells due to abnormal chromosomal segregation, disruption of DNA replication and error repair, and increased probability of somatic mutations and structural variants, which is aligned with the “unfavorable genomes” hypothesis [90].
Cryptic mosaicism in the germ layer of the gonad has been proposed as one of the
possible causes of unexplained chromosomal abnormalities in the fetus. Nevertheless, parental gonadal mosaicism should be considered when the karyotype is normal and a specific abnormality is recurrently observed [91-93]. It has been suggested that the genomic instability found in POC samples obtained from RPL families may be caused by genetic disorders that occur either during parental gametogenesis or at the postzygotic stage due to the function of the embryonic genome. Although the cause of genomic instability in many of these cases is still unknown, it is believed that some normal variants of the population carried by the maternal/paternal haplotype genome can cause genomic instability when combined in the form of the fetal genome. This is particularly
true for some large structural variants, such as CNVs, particularly in the pericentromeric and subtelomeric regions [15, 19, 94]. Despite the lack of specific phenotypic effects, these CNVs can affect the correct pairing of chromosomes in meiosis and mitosis, leading to more genomic instability and miscarriage [89].
Over the last decade, evidence gathered from analyzing CNVs in POC samples, particularly in the placenta, has shown that somatic genomic rearrangements are a characteristic of the placental genome compared to other fetal tissues. It has been postulated that these somatic rearrangements may contribute to the trophoblast invasion essential for normal implantation and placental function [95, 96]. As a hallmark of normal pregnancy in human and mouse placental genomes, somatic rearrangements primarily affect the genes responsible for cell proliferation and trophoblast invasion. Based on these findings, it has been hypothesized that the placental genomes function similarly to cancer genomes during metastatic and invasive phases, in which spontaneous structural genomic rearrangements lead to the high heterogeneity of somatic CNVs [95]. Recent evidence has shown that the load of these de novo CNVs in RPL samples is half the CNV load in uncomplicated first-trimester pregnancies with similar gestational age (first-trimester elective termination), mainly due to the lower number of de novo duplications [5, 19]. In chorionic villi samples of the RPL couples, for instance, some genes have not shown CNVs’ enrichment compared to normal pregnancies, particularly in the genes regulated by transcription factors essential in early embryonic development and regulation of fundamental cellular processes, such as E2F, ZF5, KLF4, AP2, and SREBP [90].
In addition to widespread CNVs in the placental genome, it has also been found that some pathogenic SNVs in developmentally critical genes may also predispose to recurrent cases of fetal aberrant growth. In case-control association studies conducted on the fetal genes of POC samples, the most frequently analyzed SNVs are related to immune tolerance (HLA- and KIR-genes), inflammation (IFNγ, TNF, TGF-B1, IL-genes), blood coagulation (MTHFR, F2, F5, VEGFA), and placental development and function (PAPPA, TP53, NOS3, hCGbeta locus) [5]. Recent NGS-based studies of parent-offspring trios have shed light on the number and distribution of these changes in human diseases. They have also identified factors that increase the likelihood of these changes occurring in offspring [5]. New mutations are the primary cause of early-onset genetic disorders such as mental retardation, autism spectrum disorder, and other developmental diseases [58]. Further studies are needed to determine how the genomic factors of RPL couples and their combination may affect the risk of future miscarriages and developmental disorders in live offspring.
Immunological factors involved in RPL
Currently, research on the role of immunological factors in RPL focuses mainly on autoantibodies in the mother’s blood, natural killer cells (NKCs), particularly those recruited to the decidua, serum and uterine level of cytokines, classical and non-classical HLA polymorphisms, as well as the expression of HLA antigens on the embryonic trophoblast [97-99]. The role of immunological factors in RPL can arise from two sources: 1) factors generated by the mother’s immune system and decidual microenvironment established by maternal genomic background, and 2) factors originating from fetal cells due to the maternal/paternal genomic background that constitutes the fetal genome. Given that pregnancy results from incessant and reciprocal interactions between the POC (embryo and subsequently fetus and fetal membranes) and the maternal uterus, it is challenging to distinguish these immunological factors and their genetic source.
The importance of these factors varies during different stages of embryonic development. For example, the blastocyst develops trophoblast by 5-6 days after fertilization and produces chorionic gonadotropins to provide a message for recognition of pregnancy and maintenance of progesterone production. Chorionic gonadotropins also affect the uterus locally to support implantation and persistence of pregnancy by modulating the immune cells, enhancing the decidualization of stromal cells, and promoting angiogenesis [100]. The uterine glands and their secretions also play a crucial biological role in implantation, uterus acceptance, blastocyst survival, and embryo growth. The function of these glands in the implantation site is regulated by the trophoblast (with fetal origin) and by decidual stromal cells and NKCs in the uterus microenvironment (with maternal origin). The delicate immunological balances among these key players are critical for the maintenance and persistence of pregnancy, especially during the first trimester [101].
Furthermore, recent studies have emphasized the role of uterine mesenchymal stromal/stem cells (MSCs), particularly decidual stromal cells, to facilitate decidualization in favor of pregnancy. Cumulative pieces of evidence indicate that the deregulation of decidualization due to defective endometrial stromal cells may lead to RPL. However, the essential molecular mechanisms underlying the defect of decidualization and its related pathogenesis remain unknown [102, 103].
Maternal immunological factors
Medawar first proposed mechanisms to prevent rejection of the fetus by the mother in 1953, including physical separation of maternal and fetal tissues by the placenta, immaturity of fetal antigens responsible for graft rejection, and maternal immunological tolerance, which reflects the involvement of fetal-maternal immunological factors in maintaining pregnancy and their potential role in RPL [104]. The activity of the maternal immune system regarding immune tolerance that can contribute to the occurrence of RPL depends on the maternal genomic background. Accordingly, maternal immune tolerance is associated with fetal antigen specificity, thus, the imbalance of systemic and local immune responses under special conditions can be considered as a leading player of abortion or fetal death [105]. Some of the known pathological processes in this regard include pregnancy infections (which lead to inducing macrophages to secrete high levels of the T helper 1 (Th1) cytokine and a disruptive cytokine balance at the fetal-maternal interface), fetal antigen recognition by maternal antibodies (such as the destruction of fetal erythrocytes by Rh antigen or the destruction of platelets by HPA-1 and HPA-2 antigens) [105-107].
Diagnosing immunological causes in an aborted fetus can be challenging due to the complex mother-fetal relationship. However, maternal immune regulation disorders are considered to be the leading cause of idiopathic RPL and are divided into two categories: autoimmune abnormalities and alloimmune abnormalities. Autoimmune abnormalities are further classified into two groups: maternal autoimmune diseases and the presence of autoantibodies in maternal peripheral blood. Alloimmune abnormalities depend on the presence of fetal antigens [8]. Figure 2 shows a summary of the immunological etiology of RPL in the form of a flowchart.
Autoimmune abnormalities
Various studies have confirmed the link between autoimmune abnormalities and RPL. The immunological factors responsible for RPL can be categorized into two groups: maternal autoimmune diseases and the presence of autoantibodies in the mother’s blood. Autoimmune diseases (AIDs) are chronic inflammatory conditions in which the immune system mistakenly attacks the body’s tissues, causing damage. Studies have shown that people with AID, such as APS, systemic lupus erythematosus, Sjogren’s syndrome, rheumatoid arthritis, systemic sclerosis, and undifferentiated connective tissue disease, have a higher risk of RPL [8]. Apart from the direct impact of maternal autoimmune diseases in causing miscarriage, it is also possible that these AIDs contribute to RPL by producing tissue-specific or non-tissue-specific autoantibodies [8]. Non-tissue-specific antibodies mainly comprise antiphospholipid antibodies (aPLs) and antinuclear antibodies (ANAs), while tissue-specific antibodies include anti-sperm and anti-ovary antibodies. The autoantibodies aPLs are categorized into classic and non-classic. Classical aPLs include lupus anticoagulant, anticardiolipin antibody, and anti β2 glycoprotein I antibodies (aβ2GP I) [108].
Non-classical aPLs, such as anti-β2GP I domain I antibodies, anti-prothrombin antibodies (aPT-A), and anti-phosphatidylserine/prothrombin complex antibodies (aPS/PT), have also been found to be associated with RPL [109].
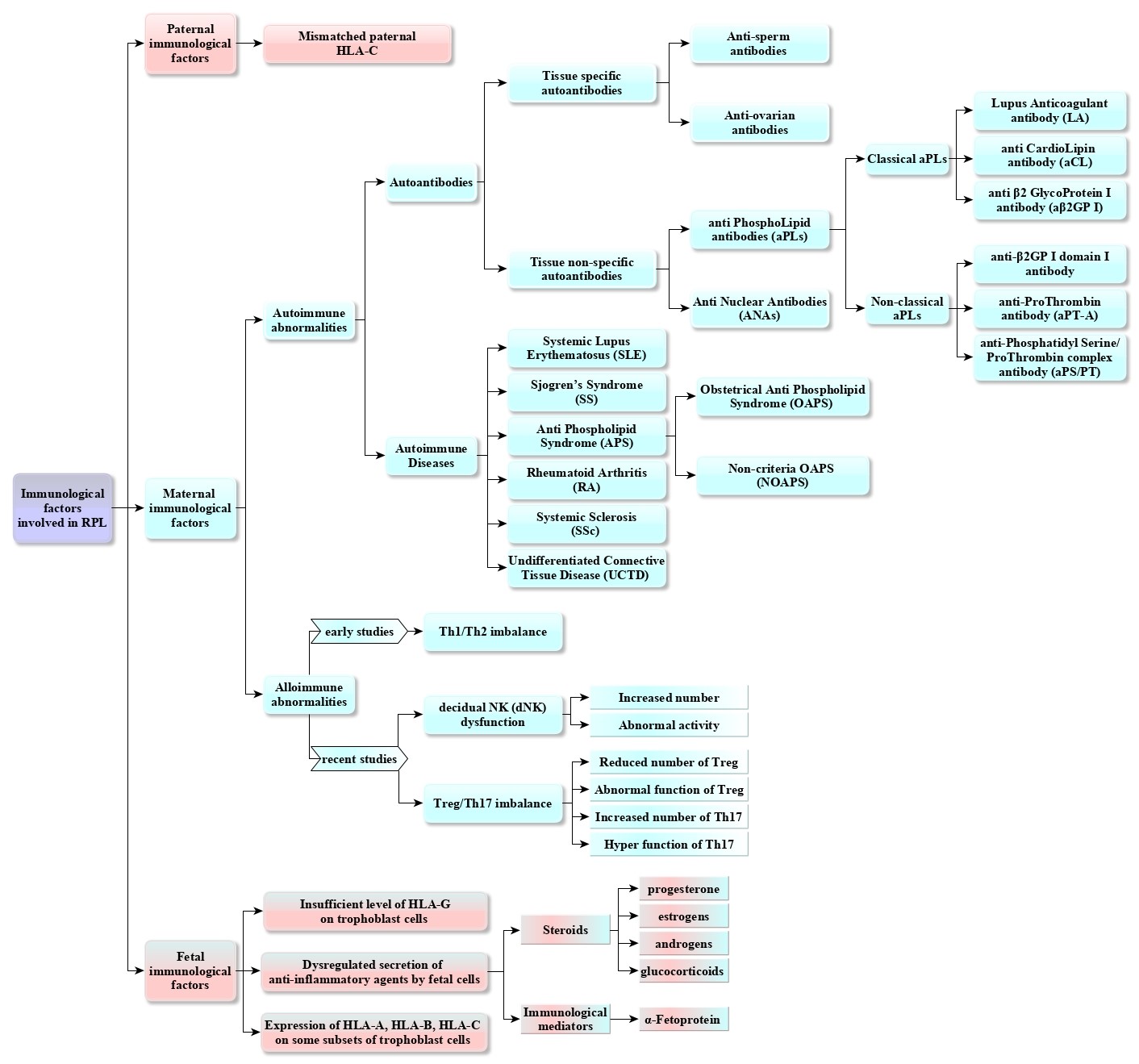
Fig. 2. The flowchart illustrates a summary of the immunological etiology of RPL.
Thrombosis, RPL, thrombocytopenia, and persistent positive serum aPLs are the key aspects of APS. Obstetrical antiphospholipid syndrome (OAPS) is the term used for APS patients with primary symptoms of pathological pregnancy, and non-criteria OAPS is used for those with typical clinical features or laboratory criteria [110].
ANAs are a group of autoantibodies that bind to DNA, RNA, protein, or molecular complexes in the nucleus [111]. More than 20 types of ANAs have been identified [112]. Studies have found that women who experience RPL often have a higher positive rate of ANAs [111]. Overall, these autoantibodies can damage the tissues of the mother’s body, the placenta, and its vascular endothelial cells, leading to pregnancy loss, in addition to their direct effect on the fetus.
Alloimmune abnormalities
Immune tolerance disorders in the maternal-fetal interface are often caused by alloimmune problems related to the presence of fetal antigens in the mother’s body. These problems are recognized as one of the main causes of RPL. Although environmental factors that affect the immune system’s function can contribute to the occurrence of such problems, the maternal genome also plays a crucial role in the pathogenesis of the disease by determining the function of the maternal immune system [113, 114]. Alloimmune problems can influence the fate of the fetus at two-time intervals. The first is related to the uterine microenvironment before implantation, while the second is associated with the complex relationship between immune cells at the maternal-fetal interface. Both environmental and maternal genome factors are involved in both time intervals.
The successful implantation of an embryo is an important part of pregnancy maintenance. The process involves various factors, including a healthy embryo with the appropriate developmental potential from the maternal and paternal genome and a suitable intrauterine environment with appropriate hormone levels that depend on both environmental factors and the maternal genome. Before implantation, the endometrium is decidualized under the combined effect of estrogen and progesterone. The endometrium has a short window period for the blastocyst to attach when it enters the uterine cavity. During this time, endometrial stromal fibroblasts differentiate, and immune cells such as uterine NKCs and macrophages begin to accumulate [115-117]. During implantation, several interleukins (ILs) are secreted by endometrial epithelial and stromal cells, including IL-6, IL-10, IL-12, IL-15, IL-18, and leukemia inhibitory factor (LIF). These ILs form an intricate network that orchestrates both the proliferation and maturation of uterine NKCs, controls the function of Treg and regulatory B cells, inhibiting the secretion of antifetal antibodies, and supports trophoblast invasion and decidua formation. Recent studies have shown that dysregulation of the ILs network compromises implantation and leads to RIF [118].
After successfully implanting the embryo, a complex network of cell-to-cell interactions between maternal and fetal cells is formed, creating the maternal-fetal interface. This process begins with embryonic trophoblasts differentiating into syncytiotrophoblasts and extravillous trophoblast cells (EVTs). An imbalance of immune hemostasis at the maternal-fetal interface can be a significant risk factor for RPL. The maternal aspect of this relationship is primarily related to the function of two types of decidual cells: decidual stromal cells (60%) and decidual immune cells (DICs) (40%). Uterine NKCs, which account for 70% of DICs, can recognize HLA-G on the surface of EVTs and activate themselves to secrete growth factors that promote embryonic development [119]. Additionally, macrophages (10-20% of DICs), T cells (10-20% of DICs), and dendritic cells all play a crucial role in maintaining immune homeostasis at the maternal-fetal interface [120]. However, the relationship between the number and function of these DICs and RPL is not yet fully understood.
Early studies have attributed alloimmune-related RPL to Th1/Th2 imbalance. Nevertheless, various mechanisms to prevent autoimmunity by distinguishing self-antigens from non-self-antigens have received more attention in recent years. Peripheral tolerance to self-antigens is mainly regulated by a subset of immunosuppressive CD4+ FOXP3+ T cells called regulatory T cells (Tregs) [121]. Many in vivo and clinical studies on pregnant women have demonstrated that maternal Treg cells develop systemically and locally at the fetal-maternal interface during pregnancy, and their constant proliferation and regulation are essential for maintaining fetal tolerance. Indeed, before embryo implantation, Treg cells accumulate in the lymph nodes of the uterus in response to hormonal fluxes and stimulation by fetal antigens [122]. Therefore, Treg cells play a role in both pre-implantation and maternal-fetal interface. The imbalance between Treg and Th17 cells has increased the risk of unexplained RPL. Studies have shown that either an increase in the number and overactivity of Th17 cells or a decrease in Treg cells’ number and abnormal function can significantly contribute to RPL [123]. While alloimmune problems have been the main area of study in RPL, the complexity of immune cells and factors responsible for the immune response means that current research is still in its early stages and requires more targeted investigations.
Paternal immunological factors
There have been limited studies investigating the role of paternal immunological factors in RPL due to the lack of connection between the fetus and the father’s body. The research in this field has mainly focused on the immunological factors inherited from the paternal haploid genome that comprise the fetal genetic pool. For instance, studies have indicated that the mismatched paternal HLA-C, the only classical HLA class-I expressed on the trophoblast, with the mother can lead to an alloimmune response. A study conducted to examine the frequency of paternal alleles of HLA-C*07 and HLA-C*17 (the most immunogenic HLA-C antigens) and the presence of HLA-specific antibodies in maternal blood revealed that HLA-C incompatibility between couples is significantly associated with unexplained RPL [124].
Recently, new findings have emerged, indicating the role of the father’s immune system health status as a result of pregnancy. This discovery has drawn attention to the role of paternal immunological factors in the occurrence of RPL. For instance, a large multicentre retrospective study (iFAME-Fertility) conducted in a Dutch population showed a significantly higher miscarriage rate in men with inflammatory arthritis [125]. These findings highlight the importance of investigating the relationship between unexplained RPL and paternal immunologic factors, including immunomodulatory factors in paternal peripheral blood and seminal fluid. Therefore, a hospital-based multicentrecase–control study and prospective cohort study (REMI III project) have been designed to examine this relationship [126].
Fetal immunological factors
The role of fetal cells in maintaining a pregnancy can be considered in two aspects: 1) A functional aspect that has been suggested for long time ago by Medawar’s strategies is the antigen-immaturity of fetal cells that enables them to escape from recognition by maternal immune system and is mostly related to the hierarchical levels of embryonic and fetal cells; 2) the endocrine and paracrine secretions of small molecules of nucleic acid, proteins, growth factors and anti-inflammatory agents from fetal cells that are mainly related to the maternal-paternal genomic background.
In the first trimester, the placenta forms an EVT structure that interacts with the mother’s immune system. This interaction can create a microenvironment that its imbalance causes immunological attacks against the fetus, leading to miscarriage [119]. Despite such close contact, in vitro studies have shown any antigenic stimulation of maternal lymphocytes by fetal trophoblastic cells [127]. The best explanation for this observation is low-expressed classical HLA in trophoblast. Despite the presence of paternal HLA alleles in the fetal genome, their expression on fetal cells and their possible role in miscarriage remains controversial. Hypothetically, this is where other genetic factors could affect the outcome of HLA involvements. For example, maternal antibodies against paternal HLA antigens have been observed in 10% to 30% of expected pregnancies. However, there is no documented abortion resulting from these antibodies since new HLA antigens (from paternal sources) expressed on fetal membranes are often tolerogenic rather than immunogenic [128].
Trophoblast cells selectively express specific non-polymorphic HLA subsets, such as HLA-G, which help protect the fetus against uterine NKC activity [119]. However, some studies have been published on natural fertility and pregnancy in individuals who lack both HLA-G alleles, which is found in different ethnicities with a prevalence of 0.6% to 11% [129]. On the other hand, it has been found that some subsets of trophoblast cells may slightly express classical HLA antigens such as HLA-A, HLA-B, or HLA-C. Consequently, their presence might be a possible cause for abortion due to the development of antibodies with high specificity to HLA antigens [130].
Another suggested approach fetal cells use to maintain pregnancy is their immunosuppressive properties. It has been revealed that fetal and placental cells can release anti-inflammatory agents as endocrine (transferred from the fetal to the maternal bloodstream) and paracrine (locally in the site of the fetal-maternal interaction) manner, leading to suppression/modulation of the maternal immune system [106]. Generally, these compounds can be divided into two groups: 1) steroids, including progesterone, estrogens, androgens, and glucocorticoids that enter the maternal bloodstream and originate from fetoplacental, adrenal, and fetal liver tissues and play a role in immunological processes related to pregnancy, from implantation to delivery [131]; 2) another group is complex protein molecules that transfer most of the immunological information from the fetus to the mother, such as α-Fetoprotein that is produced in the fetal liver and transmits the immunosuppressive message to the mother [132].
Moreover, in vitro studies have shown that fetal cells have more potential for immunosuppression than their adult counterparts. For example, studies using co-culture systems have shown that unlike adult dendritic cells, fetal ones strongly reduce the proliferation of CD8+ T cells. Additionally, these fetal cells induce Treg cells and inhibit TNF-α production in T cells, thereby preventing pro-inflammatory responses [133]. The transcriptomic investigation of adult and fetal dendritic cells revealed more than three thousand differentially expressed genes [133]. Another example is MSCs, whose notable feature is immunosuppression [134]. Numerous clinical trials have been conducted on this feature and its application in cell therapy for inflammatory diseases [135, 136]. Accordingly, many studies comparing MSCs of fetal sources with adult ones have disclosed more immunosuppressive activity of fetal tissue-derived MSCs [137, 138].
Conclusion
Based on the literature, nearly 50% of RPL cases are considered idiopathic miscarriages, and immunological factors have been suggested as one of its possible causes. The genomic similarity of aborted fetuses during RPL is the same as sibling similarity in a family, so identifying a common factor that causes RPL in a mother is very challenging. Therefore, the immunological factors involved in miscarriage can either result from the maternal genomic background or fetal origin due to the maternal-paternal genomic background. Considering that pregnancy results from the constant interaction between the pregnancy product (placenta, fetus, and fetal membranes) and the mother’s uterus, the precise determination of immunological factors and their genomic origin would be promising to develop new therapeutic approaches to help couples.
Ethical Considerations
All ethical considerations were followed in compiling this work.
Funding
This study was supported by Yazd Reproductive Sciences Institute.
Conflict of Interest
The authors declare that there is no conflict of interest regarding this article's publication and financial issues.
Acknowledgements
The authors acknowledge Dr. Nasrin Ghasemi for her support as the head of the Abortion Research Center.
Authors’ Contributions
All the authors have played an equal role in implementing this project.
References
- Jauniaux E, Farquharson RG, Christiansen OB, Exalto N. Evidence-based guidelines for the investigation and medical treatment of recurrent miscarriage. Hum Reprod. 2006; 21(9): 2216-222.
- de Ziegler D, Frydman RF. Recurrent pregnancy losses, a lasting cause of infertility. Fertil Steril. 2021; 115(3): 531-32.
- Vomstein K, Feil K, Strobel L, Aulitzky A, Hofer-Tollinger S, Kuon RJ, et al. Immunological risk factors in recurrent pregnancy loss: Guidelines Versus Current State of the Art. J Clin Med. 2021; 10(4): 322-31.
- Rull K, Nagirnaja L, Laan M. Genetics of recurrent miscarriage: challenges, current knowledge, future directions. Front Genet. 2012; 3(1): 34.
- Kasak L, Rull K, Laan M. Chapter 21 - Genetics and Genomics of Recurrent Pregnancy Loss. In: Leung PCK, Qiao J, editors. Human Reproductive and Prenatal Genetics: Academic Press; 2019. p. 463-94.
- Hoseini SM, Montazeri F, Froughmand AM, Dehghani M, Ghadimi HR. Introduction to genetic testing–applications, advantages and disadvantages. Genet 3rd millennium. 2014; 12(11): 3544-563.
- Ibrahim Y, Johnstone E. The male contribution to recurrent pregnancy loss. Translational Andrology and Urology 2018 (S1): 317-27.
- Cao C, Bai S, Zhang J, Sun X, Meng A, Chen H. Understanding recurrent pregnancy loss: recent advances on its etiology, clinical diagnosis, and management. Med Rev (Berl). 2022; 2(6): 570-89.
- Franssen MT, Korevaar JC, van der Veen F, Leschot NJ, Bossuyt PM, Goddijn M. Reproductive outcome after chromosome analysis in couples with two or more miscarriages: case-control study. BMJ 2006; 332(7544): 759-63.
- Flynn H, Yan J, Saravelos SH, Li TC. Comparison of reproductive outcome, including the pattern of loss, between couples with chromosomal abnormalities and those with unexplained repeated miscarriages. J Obstet Gynaecol Res. 2014; 40(1): 109-16.
- Ahangari N, Doosti M, Mousavifar N, Attaran M, Shahrokhzadeh S, Memarpour S, et al. Hereditary thrombophilia genetic variants in recurrent pregnancy loss. Archives of Gynecology and Obstetrics 2019; 300(3): 777-82.
- Pal AK, Ambulkar PS, Waghmare JE, Wankhede V, Shende MR, Tarnekar AM. Chromosomal aberrations in couples with pregnancy loss: A retrospective study. J Hum Reprod Sci. 2018; 11(3): 247-53.
- Turki RF, Assidi M, Banni HA, Zahed HA, Karim S, Schulten HJ, et al. Associations of recurrent miscarriages with chromosomal abnormalities, thrombophilia allelic polymorphisms and/ or consanguinity in Saudi Arabia. BMC Med Genet. 2016; 17(S 1): 69-77.
- Paththinige CS, Sirisena ND, Kariyawasam U, Dissanayake VHW. The frequency and spectrum of chromosomal translocations in a cohort of Sri Lankans. Biomed Res Int. 2019; 2019: 9797104.
- Cockwell AE, Jacobs PA, Beal SJ, Crolla JA. A study of cryptic terminal chromosome rearrangements in recurrent miscarriage couples detects unsuspected acrocentric pericentromeric abnormalities. Hum Genet. 2003; 112(3): 298-302.
- De la Fuente-Cortés BE, Cerda-Flores RM, Dávila-Rodríguez MI, García-Vielma C, De la Rosa Alvarado RM, Cortés-Gutiérrez EI. Chromosomal abnormalities and polymorphic variants in couples with repeated miscarriage in Mexico. Reprod Biomed Online 2009; 18(4): 543-48.
- Karim S, Jamal HS, Rouzi A, Ardawi MSM, Schulten HJ, Mirza Z, et al. Genomic answers for recurrent spontaneous abortion in Saudi Arabia: An array comparative genomic hybridization approach. Reprod Biol. 2017; 17(2): 133-43.
- Nagirnaja L, Palta P, Kasak L, Rull K, Christiansen OB, Nielsen HS, et al. Structural genomic variation as risk factor for idiopathic recurrent miscarriage. Hum Mutat. 2014; 35(8): 972-82.
- Kasak L, Rull K, Sõber S, Laan M. Copy number variation profile in the placental and parental genomes of recurrent pregnancy loss families. Sci Rep. 2017; 7: 45327.
- Bagheri H, Mercier E, Qiao Y, Stephenson MD, Rajcan-Separovic E. Genomic characteristics of miscarriage copy number variants. Mol Hum Reprod. 2015; 21(8): 655-61.
- Pereza N, Ostojić S, Kapović M, Peterlin B. Systematic review and meta-analysis of genetic association studies in idiopathic recurrent spontaneous abortion. Fertil Steril. 2017; 107(1): 150-59.
- Shi X, Xie X, Jia Y, Li S. Maternal genetic polymorphisms and unexplained recurrent miscarriage: a systematic review and meta-analysis. Clin Genet. 2017; 91(2): 265-84.
- Abu-Heija A. Thrombophilia and recurrent pregnancy loss: Is heparin still the drug of choice? Sultan Qaboos Univ Med J. 2014; 14(1): 26-36.
- Liu X, Chen Y, Ye C, Xing D, Wu R, Li F, et al. Hereditary thrombophilia and recurrent pregnancy loss: a systematic review and meta-analysis. Human Reproduction 2021; 36(5): 1213-229.
- Ozdemir O, Yenicesu GI, Silan F, Köksal B, Atik S, Ozen F, et al. Recurrent pregnancy loss and its relation to combined parental thrombophilic gene mutations. Genet Test Mol Biomarkers 2012; 16(4): 279-86.
- Ang KC, Bogdanova N, Markoff A, Ch'ng ES, Tang TH. Association between M2/ANXA5 haplotype and repeated pregnancy loss: A meta-analysis. Fertil Steril. 2019; 111(5): 971-81.
- Bogdanova N, Horst J, Chlystun M, Croucher PJ, Nebel A, Bohring A, et al. A common haplotype of the annexin A5 (ANXA5) gene promoter is associated with recurrent pregnancy loss. Hum Mol Genet. 2007; 16(5): 573-78.
- Rogenhofer N, Markoff A, Ennerst X, Bogdanova N, Thaler C. Maternal and paternal carriage of the annexin A5 M2 haplotype: A possible risk factor for recurrent implantation failure (RIF). J Assist Reprod Genet. 2021; 38(1): 235-42.
- Rogenhofer N, Engels L, Bogdanova N, Tüttelmann F, Markoff A, Thaler C. Paternal and maternal carriage of the annexin A5 M2 haplotype are equal risk factors for recurrent pregnancy loss: a pilot study. Fertil Steril. 2012; 98(2): 383-88.
- Papuchova H, Kshirsagar S, Xu L, Bougleux Gomes HA, Li Q, Iyer V, et al. Three types of HLA-G+ extravillous trophoblasts that have distinct immune regulatory properties. Proceedings of the National Academy of Sciences 2020; 117(27): 15772-5777.
- Ober C, Aldrich CL, Chervoneva I, Billstrand C, Rahimov F, Gray HL, Hyslop T. Variation in the HLA-G promoter region influences miscarriage rates. Am J Hum Genet. 2003; 72(6): 1425-435.
- Saxena D, Misra MK, Parveen F, Phadke SR, Agrawal S. The transcription factor Forkhead Box P3 gene variants affect idiopathic recurrent pregnancy loss. Placenta 2015; 36(2): 226-31.
- Abdukassimova M, Kanabekova P, Bauyrzhanova Z, Ukybassova T, Kaldygulova L, Imankulova B, et al. Association of human forkhead box protein 3 (FOXP3) gene polymorphisms with idiopathic recurrent pregnancy loss among Kazakhstani women. Gene. 2021; 801: 145835.
- Nasiri M, Rasti Z. CTLA-4 and IL-6 gene polymorphisms: Risk factors for recurrent pregnancy loss. Hum Immunol. 2016; 77(12): 1271-274.
- Misra MK, Mishra A, Phadke SR, Agrawal S. Association of functional genetic variants of CTLA4 with reduced serum CTLA4 protein levels and increased risk of idiopathic recurrent miscarriages. Fertil Steril. 2016; 106(5): 1115-123.
- Hayashi Y, Nishiyama T, Nakatochi M, Suzuki S, Takahashi S, Sugiura-Ogasawara M. Association of genetic variants of PD1 with recurrent pregnancy loss. Reprod Med Biol. 2018; 17(2): 195-202.
- Cho HY, Park HS, Ko EJ, Ryu CS, Kim JO, Kim YR, et al. Association of complement factor D and H polymorphisms with recurrent pregnancy loss. Int J Mol Sci. 2019; 21(1): 1-10.
- Demetriou C, Chanudet E, GOSgene, Joseph A, Topf M, Thomas AC, et al. Exome sequencing identifies variants in FKBP4 that are associated with recurrent fetal loss in humans. Human Molecular Genetics 2019; 28(20): 3466-474.
- Maddirevula S, Awartani K, Coskun S, AlNaim LF, Ibrahim N, Abdulwahab F, et al. A genomics approach to females with infertility and recurrent pregnancy loss. Human Genetics 2020; 139(5): 605-13.
- Filges I, Manokhina I, Peñaherrera MS, McFadden DE, Louie K, Nosova E, et al. Recurrent triploidy due to a failure to complete maternal meiosis II: whole-exome sequencing reveals candidate variants. Molecular Human Reproduction 2014; 21(4): 339-46.
- Quintero-Ronderos P, Mercier E, Fukuda M, González R, Suárez CF, Patarroyo MA, et al. Novel genes and mutations in patients affected by recurrent pregnancy loss. PLOS ONE 2017; 12(10): e0186149.
- Gourhant L, Bocher O, De Saint Martin L, Ludwig TE, Boland A, Deleuze JF, et al. Whole exome sequencing, a hypothesis-free approach to investigate recurrent early miscarriage. Reproductive BioMedicine Online 2021; 42(4): 789-98.
- Lee HA, Ahn EH, Kim JH, Kim JO, Ryu CS, Lee JY, et al. Association study of frameshift and splice variant polymorphisms with risk of idiopathic recurrent pregnancy loss. Mol Med Rep. 2018; 18(2): 2417-426.
- Yazdani N, Shekari Khaniani M, Bastami M, Ghasemnejad T, Afkhami F, Mansoori Derakhshan S. HLA-G regulatory variants and haplotypes with susceptibility to recurrent pregnancy loss. Int J Immunogenet. 2018; 45(4): 181-89.
- Harrison GA, Humphrey KE, Jakobsen IB, Cooper DW. A 14 bp deletion polymorphism in the HLA-G gene. Hum Mol Genet 1993; 2(12): 2200-210.
- Kim JO, Lee WS, Lee BE, Jeon YJ, Kim YR, Jung SH, et al. Interleukin-1beta -511T>C genetic variant contributes to recurrent pregnancy loss risk and peripheral natural killer cell proportion. Fertil Steril. 2014; 102(1): 206-12.
- Zidan HE, Rezk NA, Alnemr AA, Moniem MI. Interleukin-17 and leptin genes polymorphisms and their levels in relation to recurrent pregnancy loss in Egyptian females. Immunogenetics. 2015; 67(11-12): 665-73.
- Chen H, Yang X, Du J, Lu M. Interleukin-18 gene polymorphisms and risk of recurrent pregnancy loss: A systematic review and meta-analysis. J Obstet Gynaecol Res. 2015; 41(10): 1506-513.
- Kamrani A, Rahmani SA, Mosapour P, Chavoshi R. Association of IL-33 gene rs16924159 polymorphism and recurrent pregnancy loss in Iranian Azeri women. Hormone Molecular Biology and Clinical Investigation 2020; 41(4): 321-29.
- Rasti Z, Nasiri M, Kohan L. The IL-6 -634C/G polymorphism: a candidate genetic marker for the prediction of idiopathic recurrent pregnancy loss. Int J Reprod Biomed. 2016; 14(2): 103-108.
- Qaddourah RH, Magdoud K, Saldanha FL, Mahmood N, Mustafa FE, Mahjoub T, et al. IL-10 gene promoter and intron polymorphisms and changes in IL-10 secretion in women with idiopathic recurrent miscarriage. Hum Reprod. 2014; 29(5): 1025-1034.
- Magdoud K, Granados Herbepin V, Messaoudi S, Hizem S, Bouafia N, Almawi WY, et al. Genetic variation in TGFB1 gene and risk of idiopathic recurrent pregnancy loss. Mol Hum Reprod. 2013; 19(7): 438-43.
- Jang HG, Choi Y, Kim JO, Jeon YJ, Rah H, Cho SH, et al. Polymorphisms in tumor necrosis factor-alpha (-863C>A, -857C>T and +488G>A) are associated with idiopathic recurrent pregnancy loss in Korean women. Hum Immunol. 2016; 77(6): 506-11.
- Wang X, Lin Q, Ma Z, Hong Y, Zhao A, Di W, Lu P. Association of the A/G polymorphism at position 49 in exon 1 of CTLA-4 with the susceptibility to unexplained recurrent spontaneous abortion in the Chinese population. Am J Reprod Immunol. 2005; 53(2): 100-105.
- Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, et al. PD-1: PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol. 2002; 32(3): 634-43.
- Forsberg LA, Gisselsson D, Dumanski JP. Mosaicism in health and disease- clones picking up speed. Nat Rev Genet. 2017; 18(2): 128-42.
- Mohiuddin M, Kooy RF, Pearson CE. De novo mutations, genetic mosaicism and human disease. Front Genet. 2022; 13: 983668.
- Acuna-Hidalgo R, Veltman JA, Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biology. 2016; 17(1): 241.
- Rosenfeld JA, Tucker ME, Escobar LF, Neill NJ, Torchia BS, McDaniel LD, et al. Diagnostic utility of microarray testing in pregnancy loss. Ultrasound Obstet Gynecol. 2015; 46(4): 478-86.
- van den Berg MM, van Maarle MC, van Wely M, Goddijn M. Genetics of early miscarriage. Biochim Biophys Acta. 2012; 1822(12): 1951-959.
- Gu C, Li K, Li R, Li L, Li X, Dai X, et al. Chromosomal aneuploidy associated with clinical characteristics of pregnancy loss. Front Genet. 2021; 12: 667697.
- Pylyp LY, Spynenko LO, Verhoglyad NV, Mishenko AO, Mykytenko DO, Zukin VD. Chromosomal abnormalities in products of conception of first-trimester miscarriages detected by conventional cytogenetic analysis: A review of 1000 cases. J Assist Reprod Genet. 2018; 35(2): 265-71.
- Ogasawara M, Aoki K, Okada S, Suzumori K. Embryonic karyotype of abortuses in relation to the number of previous miscarriages. Fertil Steril. 2000; 73(2): 300-304.
- Goldmann JM, Veltman JA, Gilissen C. De Novo mutations reflect development and aging of the human germline. Trends in Genetics 2019; 35(11): 828-39.
- Hassold T, Hall H, Hunt P. The origin of human aneuploidy: where we have been, where we are going. Hum Mol Genet. 2007; 16(2): 203-216.
- Stanekova V, Woodman RJ, Tremellen K. The rate of euploid miscarriage is increased in the setting of adenomyosis. Hum Reprod Open. 2018; 2018(3): 1-6.
- Ottolini CS, Newnham L, Capalbo A, Natesan SA, Joshi HA, Cimadomo D, et al. Genome-wide maps of recombination and chromosome segregation in human oocytes and embryos show selection for maternal recombination rates. Nat Genet. 2015; 47(7): 727-35.
- Al-Hassan S, Hellani A, Al-Shahrani A, Al-Deery M, Jaroudi K, Coskun S. Sperm chromosomal abnormalities in patients with unexplained recurrent abortions. Arch Androl. 2005; 51(1): 69-76.
- Ramasamy R, Scovell JM, Kovac JR, Cook PJ, Lamb DJ, Lipshultz LI. Fluorescence in situ hybridization detects increased sperm aneuploidy in men with recurrent pregnancy loss. Fertil Steril. 2015; 103(4): 906-909.
- Pu Y, Yang X, Guo Y, Zhu X, Yan L, Lu S. Sperm aneuploidy and recurrent pregnancy loss: A systematic review and meta-analysis. Cogent Biology 2020; 6(1): 1759393.
- Caseiro AL, Regalo A, Pereira E, Esteves T, Fernandes F, Carvalho J. Implication of sperm chromosomal abnormalities in recurrent abortion and multiple implantation failure. Reprod Biomed Online 2015; 31(4): 481-85.
- Esquerré-Lamare C, Walschaerts M, Chansel Debordeaux L, Moreau J, Bretelle F, Isus F, et al. Sperm aneuploidy and DNA fragmentation in unexplained recurrent pregnancy loss: A multicenter case-control study. Basic Clin Androl. 2018; 28(1): 4-10.
- Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nature Reviews Genetics 2012; 13(8): 565-75.
- Faas MM, de Vos P. Mitochondrial function in immune cells in health and disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2020; 1866(10): 165845.
- Kozakiewicz P, Grzybowska-Szatkowska L, Ciesielka M, Całka P, Osuchowski J, Szmygin P, et al. Mitochondrial DNA changes in respiratory complex I genes in brain gliomas. Biomedicines 2023; 11(4): 1183.
- Kaare M, Götz A, Ulander VM, Ariansen S, Kaaja R, Suomalainen A, Aittomäki K. Do mitochondrial mutations cause recurrent miscarriage? Molecular Human Reproduction 2009; 15(5): 295-300.
- Vanniarajan A, Govindaraj P, Carlus SJ, Aruna M, Aruna P, Kumar A, et al. Mitochondrial DNA variations associated with recurrent pregnancy loss among Indian women. Mitochondrion 2011; 11(3): 450-56.
- Azadi A, Seo DJ, Jafari Sasansara H, Van Haute M. Mitochondrial DNA variations are associated with recurrent pregnancy loss. Mitochondrial DNA A DNA Mapp Seq Anal. 2018; 29(5): 674-78.
- Lewis SE, Simon L. Clinical implications of sperm DNA damage. Hum Fertil. 2010; 13(4): 201-207.
- Tan J, Taskin O, Albert A, Bedaiwy MA. Association between sperm DNA fragmentation and idiopathic recurrent pregnancy loss: A systematic review and meta-analysis. Reprod Biomed Online 2019; 38(6): 951-60.
- Agarwal S, Agarwal A, Khanna A, Singh K. Microdeletion of Y chromosome as a cause of recurrent pregnancy loss. J Hum Reprod Sci. 2015; 8(3): 159-64.
- Mojtabanezhad Shariatpanahi A, Ahmadnia H, Torkamanzehi A, Mansouri Torshizi M, Kerachian MA. Multiplex-polymerase chain reaction for detecting microdeletions in the azoospermia factor region of Y chromosome in iranian couples with non-obstructive infertility and recurrent pregnancy loss. Int J Fertil Steril. 2018; 11(4): 253-57.
- Wettasinghe TK, Jayasekara RW, Dissanayake VH. Y chromosome microdeletions are not associated with spontaneous recurrent pregnancy loss in a Sinhalese population in Sri Lanka. Hum Reprod. 2010; 25(12): 3152-156.
- Piña-Aguilar RE, Martínez-Garza SG, Kohls G, Vargas-Maciel MA, Vázquez de Lara LG, González-Ortega C, et al. Y chromosome microdeletions in Mexican males of couples with idiopathic recurrent pregnancy loss. J Obstet Gynaecol Res. 2012; 38(6): 912-17.
- Feichtinger M, Wallner E, Hartmann B, Reiner A, Philipp T. Transcervical embryoscopic and cytogenetic findings reveal distinctive differences in primary and secondary recurrent pregnancy loss. Fertil Steril. 2017; 107(1): 144-49.
- Khoury MJ, Erickson JD. Recurrent pregnancy loss as an indicator for increased risk of birth defects: a population-based case-control study. Paediatr Perinat Epidemiol. 1993; 7(4): 404-16.
- Walfisch A, Wainstock T, Segal I, Landau D, Sheiner E. 828: Maternal history of recurrent pregnancy loss increases the risk for long term paediatric neurological morbidity of the offspring. American Journal of Obstetrics & Gynecology 2017; 216(S1): 474-75.
- Davidesko S, Wainstock T, Sheiner E, Landau D, Walfisch A. Maternal history of recurrent pregnancy loss increases the risk for long-term pediatric respiratory morbidity of the offspring. Pediatr Pulmonol. 2020; 55(7): 1765-770.
- Liu P, Yuan B, Carvalho CMB, Wuster A, Walter K, Zhang L, et al. An organismal CNV mutator phenotype restricted to early human development. Cell 2017; 168(5): 830-42.
- Sõber S, Rull K, Reiman M, Ilisson P, Mattila P, Laan M. RNA sequencing of chorionic villi from recurrent pregnancy loss patients reveals impaired function of basic nuclear and cellular machinery. Scientific Reports 2016; 6(1): 38439.
- Magalhães M, Marques C, Ramos F, Jardim A, Franco S, Coelho F, et al. Why could a woman have three Trisomy 21 pregnancies? - A case report. Clin Case Rep. 2017; 5(8): 1222-225.
- Čulić V, Lasan-Trcić R, Liehr T, Lebedev IN, Pivić M, Pavelic J, Vulić R. A familial small supernumerary marker chromosome 15 associated with cryptic mosaicism with two different additional marker chromosomes derived de novo from chromosome 9: Detailed case study and implications for recurrent pregnancy loss. Cytogenet Genome Res. 2018; 156(4): 179-84.
- Hervé B, Quibel T, Taieb S, Ruiz M, Molina-Gomes D, Vialard F. Are de novo rea(21; 21) chromosomes really de novo? Clin Case Rep. 2015; 3(10): 786-89.
- Viaggi CD, Cavani S, Malacarne M, Floriddia F, Zerega G, Baldo C, et al. First-trimester euploid miscarriages analysed by array-CGH. J Appl Genet. 2013; 54(3): 353-59.
- Kasak L, Rull K, Vaas P, Teesalu P, Laan M. Extensive load of somatic CNVs in the human placenta. Scientific Reports 2015; 5(1): 8342.
- Hannibal RL, Chuong EB, Rivera-Mulia JC, Gilbert DM, Valouev A, Baker JC. Copy number variation is a fundamental aspect of the placental genome. PLoS Genet. 2014; 10(5): 1004290.
- Li D, Zheng L, Zhao D, Xu Y, Wang Y. The role of immune cells in recurrent spontaneous abortion. Reproductive Sciences 2021; 28(12): 3303-315.
- Yang X, Tian Y, Zheng L, Luu T, Kwak-Kim J. The update immune-regulatory role of pro- and anti-inflammatory cytokines in recurrent pregnancy losses. International Journal of Molecular Sciences 2023; 24(1): 132.
- Zhu L, Aly M, Kuon RJ, Toth B, Wang H, Karakizlis H, et al. Patients with idiopathic recurrent miscarriage have abnormally high TGFß+ blood NK, NKT and T cells in the presence of abnormally low TGFß plasma levels. BMC Immunology 2019; 20(1): 10.
- Makrigiannakis A, Vrekoussis T, Zoumakis E, Kalantaridou SN, Jeschke U. The role of hcg in implantation: A mini-review of molecular and clinical evidence. Int J Mol Sci. 2017; 18(6): 1305-310.
- PrabhuDas M, Bonney E, Caron K, Dey S, Erlebacher A, Fazleabas A, et al. Immune mechanisms at the maternal-fetal interface: perspectives and challenges. Nat Immunol. 2015; 16(4): 328-34.
- Ma W, Cao M, Bi S, Du L, Chen J, Wang H, et al. MAX deficiency impairs human endometrial decidualization through down-regulating OSR2 in women with recurrent spontaneous abortion. Cell and Tissue Research 2022; 388(2): 453-69.
- Shao Q, Liu X, Huang Y, Chen X, Wang H. Human decidual stromal cells in early pregnancy induce functional re-programming of monocyte-derived dendritic cells via crosstalk between G-CSF and IL-1β. Frontiers in Immunology 2020; 11.
- Hunt JS, Petroff MG, McIntire RH, Ober C. HLA-G and immune tolerance in pregnancy. FASEB J. 2005; 19(7): 681-93.
- Deshmukh H, Way SS. Immunological basis for recurrent fetal loss and pregnancy complications. Annu Rev Pathol. 2019; 14: 185-210.
- Abu-Raya B, Michalski C, Sadarangani M, Lavoie PM. Maternal immunological adaptation during normal pregnancy. Front Immunol. 2020; 11: 575197.
- Balasundaram P, Farhana A. Immunology at the maternal-fetal interface. StatPearls. Treasure Island (FL): StatPearls Publishing LLC.; 2022.
- Cohen H, Cuadrado MJ, Erkan D, Duarte-Garcia A, Isenberg DA, Knight JS, et al. 16th international congress on antiphospholipid antibodies task force report on antiphospholipid syndrome treatment trends. Lupus 2020; 29(12): 1571-593.
- Alijotas-Reig J, Esteve-Valverde E, Ferrer-Oliveras R, Sáez-Comet L, Lefkou E, Mekinian A, et al. Comparative study of obstetric antiphospholipid syndrome (OAPS) and non-criteria obstetric APS (NC-OAPS): report of 1640 cases from the EUROAPS registry. Rheumatology. 2020; 59(6): 1306-314.
- Xourgia E, Tektonidou MG. An update on antiphospholipid syndrome. Curr Rheumatol Rep. 2022; 23(12): 84.
- Liu T, Guo X, Liao Y, Liu Y, Zhu Y, Chen X. Correlation between the presence of antinuclear antibodies and recurrent pregnancy loss: A mini review. Front Endocrinol (Lausanne). 2022; 13(23): 873286.
- RPL EGGo, Bender Atik R, Christiansen OB, Elson J, Kolte AM, Lewis S, et al. ESHRE guideline: recurrent pregnancy loss. Human Reproduction Open 2018; 2018(2): 114-20.
- Tasadduq R, Ajmal L, Batool F, Zafar T, Babar A, Riasat A, Shakoori AR. Interplay of immune components and their association with recurrent pregnancy loss. Human Immunology 2021; 82(3): 162-69.
- Grimstad F, Krieg S. Immunogenetic contributions to recurrent pregnancy loss. J Assist Reprod Genet. 2016; 33(7): 833-47.
- Quenby S, Gallos ID, Dhillon-Smith RK, Podesek M, Stephenson MD, Fisher J, et al. Miscarriage matters: the epidemiological, physical, psychological, and economic costs of early pregnancy loss. Lancet 2021; 397(10285): 1658-667.
- Coomarasamy A, Gallos ID, Papadopoulou A, Dhillon-Smith RK, Al-Memar M, Brewin J, et al. Sporadic miscarriage: evidence to provide effective care. Lancet 2021; 397(10285): 1668-674.
- Coomarasamy A, Dhillon-Smith RK, Papadopoulou A, Al-Memar M, Brewin J, Abrahams VM, et al. Recurrent miscarriage: evidence to accelerate action. Lancet 2021; 397(10285): 1675-682.
- Pantos K, Grigoriadis S, Maziotis E, Pistola K, Xystra P, Pantou A, et al. The role of interleukins in recurrent implantation failure: A comprehensive review of the literature. Int J Mol Sci. 2022; 23(4): 321-26.
- Ander SE, Diamond MS, Coyne CB. Immune responses at the maternal-fetal interface. Science Immunology 2019; 4(31): 6114.
- Salvany-Celades M, van der Zwan A, Benner M, Setrajcic-Dragos V, Bougleux Gomes HA, Iyer V, et al. Three types of functional regulatory T Cells Control T cell responses at the human maternal-fetal interface. Cell Rep. 2019; 27(9): 2537-547.
- Paul WE. Self/nonself-immune recognition and signaling: A new journal tackles a problem at the center of immunological science. Self Nonself 2010; 1(1): 2-3.
- Jiang TT, Chaturvedi V, Ertelt JM, Kinder JM, Clark DR, Valent AM, et al. Regulatory T cells: new keys for further unlocking the enigma of fetal tolerance and pregnancy complications. J Immunol. 2014; 192(11): 4949-56.
- Niafar M, Samaie V, Soltani-Zangbar MS, Motavalli R, Dolati S, Danaii S, et al. The association of Treg and Th17 cells development factors and anti-TPO autoantibodies in patients with recurrent pregnancy loss. BMC Research Notes. 2023; 16(1): 302.
- Meuleman T, Haasnoot GW, van Lith JMM, Verduijn W, Bloemenkamp KWM, Claas FHJ. Paternal HLA-C is a risk factor in unexplained recurrent miscarriage. Am J Reprod Immunol. 2018; 79(2): 214-20.
- Perez-Garcia LF, Röder E, Smeele HTW, Goekoop R, Hazes JMW, Kok MR, et al. Paternal inflammatory arthritis is associated with a higher risk of miscarriage: results of a large multicentre study (iFAME-Fertility). Rheumatology 2021; 61(8): 3390-395.
- Fossé Nd, Hoorn M-Lvd, Eikmans M, Heidt S, Cessie Sl, Mulders A, et al. Evaluating the role of paternal factors in aetiology and prognosis of recurrent pregnancy loss: study protocol for a hospital-based multicentre case–control study and cohort study. BMJ Open 2019; 9(11): 33095.
- Zinkernagel RM, Hengartner H. Protective ‘immunity’ by pre-existent neutralizing antibody titers and preactivated T cells but not by so-called ‘immunological memory’. Immunol Rev. 2006; 211: 310-19.
- Lashley EE, Meuleman T, Claas FH. Beneficial or harmful effect of antipaternal human leukocyte antibodies on pregnancy outcome? A systematic review and meta-analysis. Am J Reprod Immunol. 2013; 70(2): 87-103.
- Moreau P, Dausset J, Carosella ED, Rouas-Freiss N. Viewpoint on the functionality of the human leukocyte antigen-G null allele at the fetal-maternal interface. Biol Reprod. 2002; 67(5): 1375-378.
- Apps R, Murphy SP, Fernando R, Gardner L, Ahad T, Moffett A. Human leucocyte antigen (HLA) expression of primary trophoblast cells and placental cell lines, determined using single antigen beads to characterize allotype specificities of anti-HLA antibodies. Immunology 2009; 127(1): 26-39.
- Morel Y, Roucher F, Plotton I, Goursaud C, Tardy V, Mallet D. Evolution of steroids during pregnancy: Maternal, placental and fetal synthesis. Ann Endocrinol. 2016; 77(2): 82-9.
- Stern C, Schwarz S, Moser G, Cvitic S, Jantscher-Krenn E, Gauster M, et al. Placental endocrine activity: Adaptation and disruption of maternal glucose metabolism in pregnancy and the influence of fetal sex. Int J Mol Sci. 2021; 22(23): 18-26.
- McGovern N, Shin A, Low G, Low D, Duan K, Yao LJ, et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature 2017; 546(7660): 662-66.
- Hoseini S, Montazeri F, Kalantar S, Bahrami A, Zarein F, Moghadam matin M. Mesenchymal stem cells: interactions with immune cells and immunosuppressive-immunomodulatory properties. The Scientific Journal of Iranian Blood Transfusion Organization 2020; 17(2): 147-69.
- Hoseini SM, Moghaddam-Matin M, Bahrami AR, Montazeri F, Kalantar SM. Human amniotic fluid stem cells: general characteristics and potential therapeutic applications. The Journal of Shahid Sadoughi University of Medical Sciences 2021; 28(12): 3252-275.
- Hoseini SM, Sheikhha MH, Kalantar SM, Matin MM, Aflatoonian B, Bahrami AR, et al. A comparative analysis of immunomodulatory genes in two clonal subpopulations of CD90+ amniocytes isolated from human amniotic fluid. Placenta 2020; 101: 234-41.
- Yu Y, Valderrama AV, Han Z, Uzan G, Naserian S, Oberlin E. Human fetal liver MSCs are more effective than adult bone marrow MSCs for their immunosuppressive, immunomo-dulatory, and Foxp3+ T reg induction capacity. Stem Cell Research & Therapy 2021; 12(1): 138.
- Chen PM, Yen ML, Liu KJ, Sytwu HK, Yen BL. Immunomodulatory properties of human adult and fetal multipotent mesenchymal stem cells. Journal of Biomedical Science 2011; 18(1): 49.
Type of Study:
Research |
Subject:
Immunology Received: 2023/09/8 | Accepted: 2023/12/26 | Published: 2023/12/1