Sex chromosomes usually determine the factual gender; in mammals, most females are characterized by XX chromosomes and men by XY. The Y chromosome contains particular genes that play an important role in masculinizing the fetus and secrete two significant hormones, the first anti-Müllerian duct hormone (AMH) and the second testosterone, which are essential for secondary sexual characteristics [1]. In rare genetic cases, People are phenotypically female while they have a masculine genotype (XY) [2]. This inconsistency between the phenotype and genotype is included in the group of clinical conditions called disorders of sex development (DSDs) [3]. DSDs can arise from defects with chromosomal development, gonadal function, and androgen synthesis, leading to disorders with masculinization of the external genitalia and phenotypic sex. Based on the chromosomal cases, DSDs comprised three constituents: 46, XX DSD; 46, XY DSD; and sex chromosomal DSD. XY DSD is uncommon, but it can manifest at any stage in the sexual differentiation pathway, an intricate progression involving gene expression, androgen production, and hormonal control through the binding of ligands to their respective receptor [4]. The clinical identification of 46, XY DSD in patients is typically done during investigations into delayed puberty or primary amenorrhea [5, 6]. However, our case study (DSD female) not only had a regular period cycle but also became pregnant and gave birth to a child who unfortunately died after nine months from liver disease. The early death of the first child prompted the mother to undergo conventional genetic testing, including cytogenetics and molecular genetics, which ascertained her abnormal karyotype.
This report illustrates an unprecedented case of a fertile woman with an XY chromosome who assumed that she was a chimera as a result of Twin-to-twin transfusion syndrome [7]. We report features of gonads and sex-determining organs to identify how the patient could have healthy female genitals without testis formation and also experienced two unassisted pregnancies. Whereas she would be infertile if the Y chromosome encoded sex-determining factors. It is thought to be caused by mutations in influential genes on the Y chromosome, such as point mutations or frameshifts in the Sox9 and SRY genes [8].
This study aims to show the normal shape of women’s reproductive organs and the natural conditions for a healthy pregnancy. On the other hand, genotypic problems routinely delay or completely disrupt the pregnancy process. However, in this particular case, due to the special genotypic conditions and contamination of the woman’s blood with her brother’s blood during the fetal period, the pregnancy goes through its normal course.
Case Report
A 22-year-old Caucasian mother presented to our Cytogenetic Department of Imam Khomeini Hospital for prenatal genetic screening and diagnostic tests for a secondary pregnancy. Her weight was 63 kg, height was 167 cm, and body mass index was 22.6 kg/m2. The patient had Areolar Mound breast (Tanner breast stage 4) and Mid-Escutcheon pubic hair (Tanner pubic hair stage 4) [9]. The patient was subsequently raised physically and mentally as a woman, and the female external genitalia organ was examined; she had no anatomical defects such as clitoral enlargement or other signs of virilization. She reached spontaneous menarche at the age of 11 yr and had a regular menstrual cycle. She became unassisted pregnant once and gave birth to a baby, but her daughter died nine months after birth due to liver disease.
Fertilization and egg formation significantly confirm the existence of the oviduct, uterus, and vagina with fallopian tubes with normal appearance. Abdominal and pelvic ultrasound for the first child was performed on the eleventh day of the last menstrual period, which showed that the uterus had normal size, measuring about 73mm in length and 38 mm in width, and The endometrium was regular and triple line with a thickness of 7 mm. The size, shape, and echo pattern of the right and left ovaries are 30 mm×22 mm and 40 mm×24 mm, respectively, which is mostly normal. The average diameter of the follicle in the left ovary was 15 mm by transvaginal ultrasound. Also, in the transvaginal ultrasound of the 11th week of pregnancy, the cervix was closed, and its length was 33 mm [10] (Fig. 1).
Pathology laboratory documented that the endocervical zone cells are present and negative for intraepithelial lesion or malignancy.
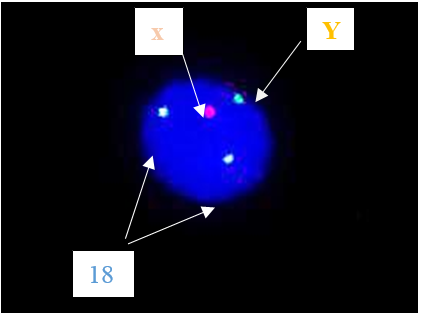
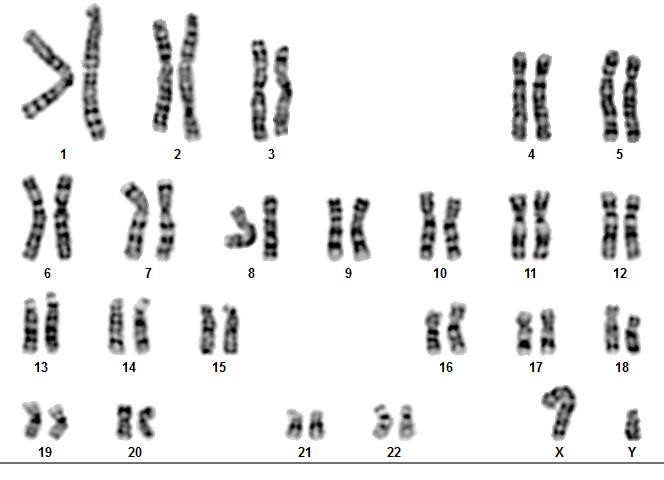
Laboratory evaluation demonstrated an average level of serum follicle-stimulating hormone (FSH) (5.8 mIU/mL), and prolactin was 29.2 ng/mL (normal 5.18 ~ 26.53 ng/mL). In addition, anti-mullerian hormone (AMH) was 2.11 ng/ml, which was within the normal range for women (1.54 - 3.5), which indicates that ovarian reserve is expected compared to women of this age Fluorescence in-situ hybridization (FISH) was obtained on an uncultured blood sample by using a set of centromeric probes (MetaSystems, XA X/Y/18 Aneusomy Probe), that found that the woman has a male chromosome aberrations karyotype (XY) (Fig. 2) [11]. We conducted additional investigations, which involved performing another cytogenetic analysis, such as GTG-banding and quantitative fluorescent polymerase chain reaction (QF-PCR) analysis of chromosomes 13, 18, 21, X, and Y. The 46, XY karyotype was revealed in 60 metaphase chromosomes from peripheral blood, which our experts analyzed using G-banding (Fig. 3) [12].
The entity of the SRY gene was confirmed through QF-PCR assay by using the Y chromosome SRY marker [13]. The SRY-box is located on the short distal arm of the Y chromosome (Yp11.3). There is plenty of evidence to suggest that this gene encodes the human testis-determining factor [1, 14]. One year after her initial presentation, she experienced a second gestational, but we still do not have valid information about the second expectancy.
Discussion
The genetic sex of individuals is determined during the embryonic stage when the reproductive system’s sexual differentiation occurs. At this point, the genital ridge can differentiate into a testis or an ovary, depending on whether the SRY gene is present. Any mutations (deletion or translocation) of the SRY gene can be accompanied by preventing the binding of SRY proteins with DNA [15, 16]. Certain genes in the testis have SRY-binding sites in their promoters or enhancers [17, 18]. When SRY binds to these sites, it triggers the formation of the testes by activating AMH [19]. In the current research, our case has a positive SRY gene on the Y chromosome without any male external genitalia. Instead, she has a normal ovarian cortex and underdeveloped female external genitalia. It shows that additional genes are responsible for the testis-determining pathway beyond SRY.
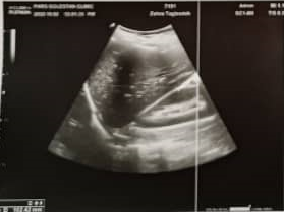
Fig. 1. Transvaginal Ultrasound of first child
Fig. 2. Fluorescence In Situ Hybridization was performed by applying the XA probe kit manufactured by Meta Systems company. One X and one Y signal were detected in 30 cells.
Fig. 3. 60 metaphase spreads were studied on the basis of the GTG technique at 500-550 band resolution
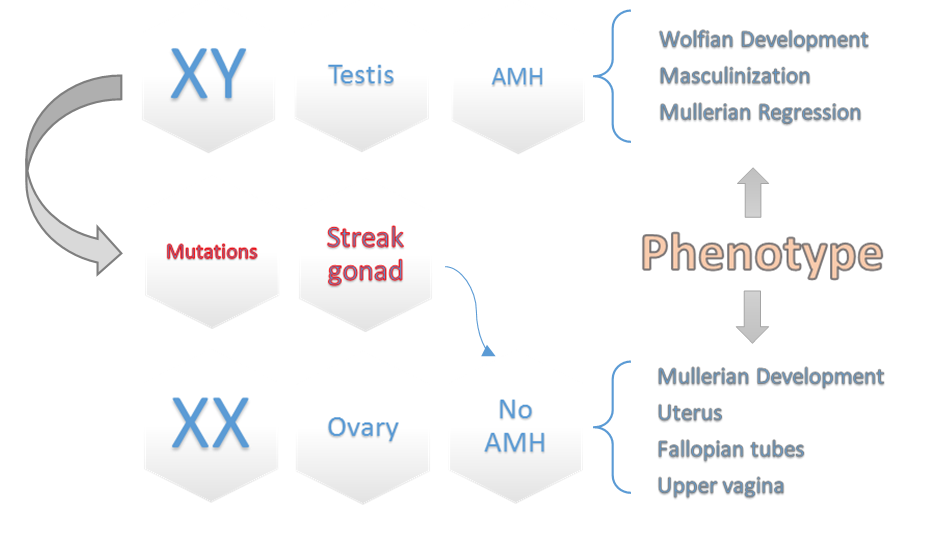
Fig.4. A schematic diagram of different pathways of sexual development interacted with AMH.
Some of these genes include SOX9, DAX1, WT-1, and SF1, which play a role in either regulating SRY or activating downstream transcription of SRY for testis-determining pathway, as well as abnormalities with the SRY protein can also lead to issues with the secretion of Testosterone and AMH (secondary sexual characteristics) [20]. This can lead to the Wolffian duct not developing into the male reproductive tract. On the other hand, without the Müllerian-inhibiting factor or androgen action, the Müllerian duct can differentiate into the oviducts, uterus, cervix, and upper vagina (Fig. 4) [21]. With the medical examinations and counseling performed by the geneticist specialist in our department, it was found that the lady had a twin brother who died during the intrauterine period, and the blood supply between the two fetuses has been shared; it can be assumed that she is a chimera by feto–fetal transfusion during pregnancy [7]. The term “chimera” refers to an organism that contains cells from two or more zygotes. There are three types of chimerism: artificial, tetragametic, and twin. Artificial chimerism can occur when transfused blood stem cells are received or through intrauterine, allogenic bone marrow, or organ transplantation. Tetragametic chimerism occurs when two spermatozoa fertilize two oocytes and fuse together [22]. Although rare in humans, tetragametic chimerism can lead to true hermaphroditism. Twin chimerism, once considered rare in humans, has been detected in 8% of twin pairs and 21% of triplets using a fluorescence technique to detect blood group chimerism. Oocyte cytogenetic analysis was not feasible, whereas oocytes are expected to be derived from 46, XX cells. Because in cases of FFT, chimerism is typically limited to blood cells [23].
The pure gonadal dysgenesis syndrome has an incidence of roughly 5 cases per 100,000 newborns. Mutations in the SRY gene are responsible for approximately 10% to 20% of cases of pure gonadal dysgenesis. This causes the retention of Mullerian ducts in individuals who are genetically male. Other genes (ZFY, SOX9, SF1, WT1, DYZ1, DAX1) are also possible contributors to this syndrome. In individuals with complete 46, XY gonadal dysgenesis, the external genitalia and Müllerian structures typically exhibit female characteristics when the gonads are dysgenetic and nonfunctional [24]. These individuals rarely experience spontaneous pubertal development, and successful pregnancy is an even rarer occurrence. However, a few cases of prosperous pregnancies through oocyte donation have been documented in patients with 46, XY gonadal dysgenesis [25, 26].
This patient was growing up as a female for 22 years regardless of her genotypic sex being male. Based on the history of the previous newborn, at the suggestion of Obstetricians and Gynecologists, she gave cytogenetic tests for the subsequent gestation. We found out her actual genotype following those examinations. According to the hypothesis of our experts, this atypical development sex occurred by FFT during the fetal period, and afterward, she became a 46 XY female chimerism with unexpected fertile ability. Due to the rarity of DSDs, the Prophylactic gonadectomy is intended for Female patients with 46 XY karyotypes since they are predisposed to gonadal malignancy [27]. The prophylactic gonadectomy was performed after examinations of immunohistochemistry, hormonal profile, ultrasound examination, molecular index, and cytogenetic analysis of the patient. Besides, these cases ought to be handled at specialized hospitals with expertise in diagnosis and treatment, along with a team of experts who can provide medical and surgical assistance. Skilled professionals should sensitively inform patients of their diagnosis, and psychological therapy should be easily accessible for patients and their families [28]. The results of this research showed that blood transfusion through the umbilical cord during the fetal period can cause this complication. This incident has caused her to become a chimera with XY karyotype. Therefore, a fetal karyotype test (CVS / Amniocentesis) is recommended to prevent this condition.
Ethical Consideration
The examined patient entered this study with her consent and in a fully informed manner, and the patient's written consent is available in the Genetics Department of Imam Khomeini Hospital Complex.
Funding
All expense for this case study is funded by the Genetics Department and Research Center of Imam Hospital Complex.
Conflict of Interests
The authors of this study have no competitive conflicts with each other.
Acknowledgments
We appreciate and thank all the staff of the genetics department and the infertility research center who have cooperated in this study, as well as the examined patient.
Authors’ Contributions
A.SF, F.N and N.R: conceptualization, experimental set-up, data interpretation, manuscript writing, and figure preparation. M.M: experimental work, data analysis, and interpretation. A.SF, F.N: data analysis, interpretation, manuscript, and figure formatting. All authors reviewed and approved the final version of the manuscript.
References
[1]. Gilbert SF. Chromosomal sex determination in mammals. Developmental Biology. 6th ed: Sinauer Associates; 2000. p. 463-66.
[2]. Hughes IA. Disorders of sex development: A new definition and classification. Best Pract Res Clin Endocrinol Metab. 2008; 22(1): 119-34.
[3]. Sudik R, Jakubiczka S, Nawroth F, Gilberg E, Wieacker PF. Chimerism in a fertile woman with 46,XY karyotype and female phenotype: Case report. Human Reproduction 2001; 16(1): 56-8.
[4]. Guidozzi F, Ball J, Spurdle A. 46,XY pure gonadal dysgenesis (Swyer-James syndrome)-Y or Y not?: A review. Obstet Gynecol Surv. 1994; 49(2): 138-46.
[5]. Michala L, Creighton SM. The XY female. Best Pract Res Clin Obstet Gynaecol. 2010; 24(2): 139-48.
[6]. Bumbulienė Ž, Bužinskienė D, Banuškevičienė G, Šidlovska E, Preikšaitienė E, Utkus A. Challenges in the diagnosis of XY differences of sexual development. Medicina (Kaunas). 2022; 58(12): 1121-132.
[7]. Mannaerts D, Muys J, Blaumeiser B, Jacquemyn Y. A rare cause of primary amenorrhoea, the XY female with gonadal dysgenesis. BMJ Case Rep. 2015; 2(7): 206609.
[8]. Sekido R, Lovell-Badge R. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature 2008;453(7197): 930-34.
[9]. Avila JT. Normal adolescent growth and development. In: Halpern-Felsher B, editor. Encyclopedia of Child and Adolescent Health. Oxford: Academic Press; 2023. p. 735-45.
[10]. Carvalho MH, Bittar RE, Brizot ML, Maganha PP, Borges da Fonseca ES, Zugaib M. Cervical length at 11-14 weeks’ and 22-24 weeks’ gestation evaluated by transvaginal sonography, and gestational age at delivery. Ultrasound Obstet Gynecol. 2003; 21(2): 135-39.
[11]. Tepperberg J, Pettenati MJ, Rao PN, Lese CM, Rita D, Wyandt H, et al. Prenatal diagnosis using interphase fluorescence in situ hybridization (FISH): 2-year multi-center retrospective study and review of the literature. Prenat Diagn. 2001; 21(4): 293-301.
[12]. Remya RS, Hariharan S, Keerthi V, Gopakumar C. Preprocessing G-banded metaphase: towards the design of automated karyotyping. SN Applied Sciences 2019; 1(12): 1123-132.
[13]. Badenas C, Rodríguez-Revenga L, Morales C, Mediano C, Plaja A, Pérez-Iribarne MM, et al. Assessment of QF-PCR as the first approach in prenatal diagnosis. J Mol Diagn. 2010; 12(6): 828-34.
[14]. Hawkins JR. Mutational analysis of SRY in XY females. Human Mutation 1992; 2(5): 347-50.
[15]. Du X, Zhang X, Li Y, Han Y. 46,XY female sex reversal syndrome with bilateral gonadoblastoma and dysgerminoma. Exp Ther Med. 2014; 8(4): 1102-104.
[16]. Jäger RJ, Anvret M, Hall K, Scherer G. A human XY female with a frame shift mutation in the candidate testis-determining gene SRY. Nature 1990; 348(6300): 452-54.
[17]. Sukumaran A, Desmangles JC, Gartner LA, Buchlis J. Duplication of dosage sensitive sex reversal area in a 46, XY patient with normal sex determining region of Y causing complete sex reversal. J Pediatr Endocrinol Metab. 2013; 26(7-8): 775-79.
[18]. Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 1990; 346(6281): 240-44.
[19]. Haqq CM, King CY, Ukiyama E, Falsafi S, Haqq TN, Donahoe PK, et al. Molecular basis of mammalian sexual determination: activation of müllerian inhibiting substance gene expression by SRY. Science 1994; 266(5190): 1494-500.
[20]. Domenice S, Corrêa RV, Costa EMF, Nishi MY, Vilain E, Arnhold IJP, Mendonca BB. Mutations in the SRY, DAX1, SF1 and WNT4 genes in Brazilian sex-reversed patients. Brazilian Journal of Medical and Biological Research 2004; 1(1): 37-44.
[21]. Josso N, Rey RA. What does AMH tell us in pediatric disorders of sex development? Front Endocrinol (Lausanne). 2020; 11(4): 619-26.
[22]. van Dijk BA, Boomsma DI, de Man AJ. Blood group chimerism in human multiple births is not rare. Am J Med Genet. 1996; 61(3): 264-68.
[23]. Binkhorst M, de Leeuw N, Otten BJ. A healthy, female chimera with 46,XX/46,XY karyotype. Journal of Pediatric Endocrinology and Metabolism 2009; 22(1): 97-102.
[24]. Jorgensen PB, Kjartansdottir KR, Fedder J. Care of women with XY karyotype: A clinical practice guideline. Fertil Steril. 2010; 94(1): 105-13.
[25]. Chen MJ, Yang JH, Mao TL, Ho HN, Yang YS. Successful pregnancy in a gonadectomized woman with 46,XY gonadal dysgenesis and gonadoblastoma. Fertil Steril. 2005; 84(1): 217-24.
[26]. Dumic M, Lin-Su K, Leibel NI, Ciglar S, Vinci G, Lasan R, et al. Report of fertility in a woman with a predominantly 46,XY karyotype in a family with multiple disorders of sexual development. J Clin Endocrinol Metab. 2008; 93(1): 182-89.
[27]. Liu AX, Shi HY, Cai ZJ, Liu A, Zhang D, Huang HF, et al. Increased risk of gonadal malignancy and prophylactic gonadectomy: A study of 102 phenotypic female patients with Y chromosome or Y-derived sequences. Human Reproduction 2014; 29(7): 1413-419.
[28]. Basri NI, Soon CH, Ali A, Ghani NAA, Zainuddin AA. Prophylactic gonadectomy in 46 XY females; why, where and when? Hormone Molecular Biology and Clinical Investigation 2021; 42(3): 325-28.