International Journal of Medical Laboratory
Shahid Sadoughi University of Medical Sciences
Fri, Jun 26, 2026
[Archive]

Volume 11, Issue 4 (November 2024)
IJML 2024, 11(4): 274-282 |
Back to browse issues page


Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Astani A, Khalili A. Inborn Errors of Immunity Associated with Herpes Simplex Encephalitis. IJML 2024; 11 (4) :274-282
URL: http://ijml.ssu.ac.ir/article-1-552-en.html
URL: http://ijml.ssu.ac.ir/article-1-552-en.html


Hematology and Oncology Research Center, Noncommunicable Diseases Research Institute, Shahid Saduoghi University of Medical Sciences, Yazd, Iran & Department of pediatrics, Shahid Sadoughi Hospital, School of Medicine, Shahid Sadoughi University of Medical Sciences, Yazd, Iran, Children Growth Disorder Research Center, Shahid Sadoughi University of Medical Sciences, Yazd, Iran
Full-Text [PDF 241 kb]
(425 Downloads)
| Abstract (HTML) (569 Views)
References
Full-Text: (249 Views)
Introduction
Inborn errors of immunity (IEI) are heterogeneous disorders characterized by genetic defects in one or more components of the immune system. Patients with IEI are susceptible to frequent bacterial, fungal, and viral infections. They are prone to specific microorganisms according to the type of IEI. Herpes simplex virus is a common pathogen in humans that can disseminate and present with herpes encephalitis in specific primary immune deficiency [1].
The most common cause of encephalitis is viral infections. Herpes simplex type 1, 2 (HSV1, 2) is the most common and important cause of viral encephalitis. Herpes simplex encephalitis (HSE) represents five to fifteen percent of infectious encephalitis [2]. The incidence of HSE is around 1-2/250,000 per year [3]. They are responsible for significant mortality and morbidity and neurologic complications [4]. Innate and acquired immunity have a central role in controlling HSV infection. However, the herpes virus has developed mechanisms that enable it to evade the immune system. Sometimes immunologic responses to HSV contribute to the pathogenesis of disease, as seen in HSV keratitis and encephalitis. Genetic polymorphisms can also predispose people to severe herpes infections [5]. HSV is a neuroinvasive pathogen. It can lead to cytotoxicity and neurologic deficit. In HSV encephalitis, direct viral invasion and immune-mediated disease may coexist [6]. The most common site of brain involvement is the fronto-temporal region. HSV encephalitis is a rare presentation of HSV-infected individuals. And there are no epidemics of HSE. High frequencies of consanguinity in parents of children with HSE have been reported. These findings strongly suggest that HSE has a genetic susceptibility at least in some people [7]. Based on a previous study, HSV encephalitis may occur in several primary immunodeficiencies [8]. There is incomplete evidence about the increased susceptibility to HSE in HSV-infected patients. Several studies have shown that defects in Toll-like receptors (TLR)3 signaling and interferon (IFN) production and IFN-independent cell-intrinsic mechanisms contribute to HSE [3]. In the present review, we discuss primary immunodeficiencies and genetic abnormalities that predispose individuals to HSE.
TLR mutation
The innate immune system is the first defense mechanism against microorganisms. Pattern recognition receptors (PRRs), such as TLRs, are important elements in the innate immune system. Pattern recognition receptors can activate a signaling cascade that leads to the production of anti-viral and pro-inflammatory cytokines. TLRs can recognize certain membrane molecules, such as lipoproteins, lipids, and proteins. TLR3 recognizes viral dsRNA [9, 10]. TLR3 initiates upregulation and expression of INF α/β and activates anti-viral protein synthesis. It is entirely dependent on Toll-interleukin-1 receptor (TIR)-domain-containing adaptor-inducing IFN-β (TRIF). TLR3 is found on immunologic cells (myeloid dendritic cells, mast cells, and macrophages), neurons, epithelial cells, and fibroblasts. Dendritic cells are one of the most important cells that express TLR3 and contribute to anti-viral immunity by production of INFα/β. TLR3 facilitates antigen presentation in dendritic cells, thereby inducing lymphocyte-mediated immunologic responses [11]. The intact TLR3 signaling pathway is important for effective anti-viral immunity in HSV infection. However, impairment in the signaling cascade may lead to HSE. The anti-viral responses in HSV infection are dependent on TLR3-mediated INFα, INFβ, INFλ, and INFγ production [7, 12, 13].
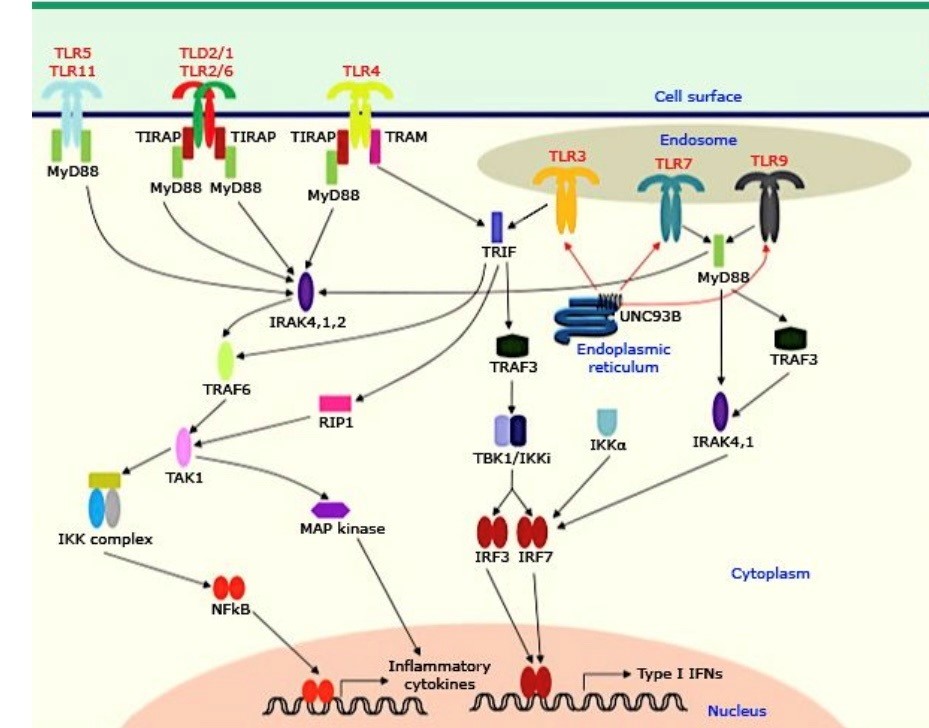
Discovery of autosomal dominant and autosomal recessive TLR3 mutations in patients with HSE confirms that childhood HSE has a genetic basis, and the TLR3 -INF signaling pathway plays a crucial role in immunity against HSV infection in some children. Investigations showed that other molecules in this signaling pathway may be involved in the pathogenesis of HES in some children. Autosomal recessive TLR3 deficiency is characterized by a complete lack of TLR3 and is accompanied by central nervous system (CNS) infection by HSV. The patients with TLR3 deficiency are not susceptible to HSV-1 infection outside the CNS, even during the clinical course of HSE. Evidence suggests that TLR3-independent dsRNA-responsive pathways contribute to controlling HSV-1 infection outside the CNS in TLR3-deficient patients [14]. Impaired production of INF in infected neurologic cells results in cell death and presentation of clinical signs and symptoms of HSE. Reactivation of latent herpes virus and inadequate control of HSV in the brain may lead to HSE and relapse of the disease. Immune reaction may persist up to 10 years after the beginning of HSE. There is a hypothesis that the recurrence of HSE in the case of primary immunodeficiency is related to a TLR3 defect [15]. It seems that 66% of patients with a defect in the TLR3 pathway develop relapses of HSE. A recent investigation showed that 27% of patients with HES progress to autoimmune encephalitis in several weeks after the onset of HSE [16]. The importance of the TLR3 signaling pathway in controlling HSE was confirmed by the finding of defects in other molecules that have a significant role in this pathway [17] (Fig. 1).
UNC93B1 mutation
UNC93B is a transmembrane molecule that has an important role in nucleic acid-sensing TLR signaling. It regulates TLR7/9 function by transferring them into the endolysosomes [18]. In the neural cells, TLR3 is in the endoplasmic reticulum. It is associated with the UNC91B protein. The association of TRL3 and UNC93B occurs over an extended period. After activation and participation of UNC93B, TLR3 is transported into endosomes. A mutation in UNC93B can cause the prevention of signaling in TLR3/7/9 pathways. Autosomal recessive deficiency in the UNC93B gene led to unresponsiveness of the TLR3 signaling pathway, and the patient became prone to HSE [15]. UNC93B protein is important for two reasons: first, it is very critical for host defense against HSV infection, and second, is that UNC93B has an essential role in nucleic acid-sensing TLR function [19].
The most common cause of encephalitis is viral infections. Herpes simplex type 1, 2 (HSV1, 2) is the most common and important cause of viral encephalitis. Herpes simplex encephalitis (HSE) represents five to fifteen percent of infectious encephalitis [2]. The incidence of HSE is around 1-2/250,000 per year [3]. They are responsible for significant mortality and morbidity and neurologic complications [4]. Innate and acquired immunity have a central role in controlling HSV infection. However, the herpes virus has developed mechanisms that enable it to evade the immune system. Sometimes immunologic responses to HSV contribute to the pathogenesis of disease, as seen in HSV keratitis and encephalitis. Genetic polymorphisms can also predispose people to severe herpes infections [5]. HSV is a neuroinvasive pathogen. It can lead to cytotoxicity and neurologic deficit. In HSV encephalitis, direct viral invasion and immune-mediated disease may coexist [6]. The most common site of brain involvement is the fronto-temporal region. HSV encephalitis is a rare presentation of HSV-infected individuals. And there are no epidemics of HSE. High frequencies of consanguinity in parents of children with HSE have been reported. These findings strongly suggest that HSE has a genetic susceptibility at least in some people [7]. Based on a previous study, HSV encephalitis may occur in several primary immunodeficiencies [8]. There is incomplete evidence about the increased susceptibility to HSE in HSV-infected patients. Several studies have shown that defects in Toll-like receptors (TLR)3 signaling and interferon (IFN) production and IFN-independent cell-intrinsic mechanisms contribute to HSE [3]. In the present review, we discuss primary immunodeficiencies and genetic abnormalities that predispose individuals to HSE.
TLR mutation
The innate immune system is the first defense mechanism against microorganisms. Pattern recognition receptors (PRRs), such as TLRs, are important elements in the innate immune system. Pattern recognition receptors can activate a signaling cascade that leads to the production of anti-viral and pro-inflammatory cytokines. TLRs can recognize certain membrane molecules, such as lipoproteins, lipids, and proteins. TLR3 recognizes viral dsRNA [9, 10]. TLR3 initiates upregulation and expression of INF α/β and activates anti-viral protein synthesis. It is entirely dependent on Toll-interleukin-1 receptor (TIR)-domain-containing adaptor-inducing IFN-β (TRIF). TLR3 is found on immunologic cells (myeloid dendritic cells, mast cells, and macrophages), neurons, epithelial cells, and fibroblasts. Dendritic cells are one of the most important cells that express TLR3 and contribute to anti-viral immunity by production of INFα/β. TLR3 facilitates antigen presentation in dendritic cells, thereby inducing lymphocyte-mediated immunologic responses [11]. The intact TLR3 signaling pathway is important for effective anti-viral immunity in HSV infection. However, impairment in the signaling cascade may lead to HSE. The anti-viral responses in HSV infection are dependent on TLR3-mediated INFα, INFβ, INFλ, and INFγ production [7, 12, 13].
Discovery of autosomal dominant and autosomal recessive TLR3 mutations in patients with HSE confirms that childhood HSE has a genetic basis, and the TLR3 -INF signaling pathway plays a crucial role in immunity against HSV infection in some children. Investigations showed that other molecules in this signaling pathway may be involved in the pathogenesis of HES in some children. Autosomal recessive TLR3 deficiency is characterized by a complete lack of TLR3 and is accompanied by central nervous system (CNS) infection by HSV. The patients with TLR3 deficiency are not susceptible to HSV-1 infection outside the CNS, even during the clinical course of HSE. Evidence suggests that TLR3-independent dsRNA-responsive pathways contribute to controlling HSV-1 infection outside the CNS in TLR3-deficient patients [14]. Impaired production of INF in infected neurologic cells results in cell death and presentation of clinical signs and symptoms of HSE. Reactivation of latent herpes virus and inadequate control of HSV in the brain may lead to HSE and relapse of the disease. Immune reaction may persist up to 10 years after the beginning of HSE. There is a hypothesis that the recurrence of HSE in the case of primary immunodeficiency is related to a TLR3 defect [15]. It seems that 66% of patients with a defect in the TLR3 pathway develop relapses of HSE. A recent investigation showed that 27% of patients with HES progress to autoimmune encephalitis in several weeks after the onset of HSE [16]. The importance of the TLR3 signaling pathway in controlling HSE was confirmed by the finding of defects in other molecules that have a significant role in this pathway [17] (Fig. 1).
UNC93B1 mutation
UNC93B is a transmembrane molecule that has an important role in nucleic acid-sensing TLR signaling. It regulates TLR7/9 function by transferring them into the endolysosomes [18]. In the neural cells, TLR3 is in the endoplasmic reticulum. It is associated with the UNC91B protein. The association of TRL3 and UNC93B occurs over an extended period. After activation and participation of UNC93B, TLR3 is transported into endosomes. A mutation in UNC93B can cause the prevention of signaling in TLR3/7/9 pathways. Autosomal recessive deficiency in the UNC93B gene led to unresponsiveness of the TLR3 signaling pathway, and the patient became prone to HSE [15]. UNC93B protein is important for two reasons: first, it is very critical for host defense against HSV infection, and second, is that UNC93B has an essential role in nucleic acid-sensing TLR function [19].
Fig. 1. Toll-like receptors: organization and signaling pathway [20]
The investigations have shown that UNC93B deficiency may cause sporadic HSE by impairment of type 1 and 3 IFN responses after viral infections. In one study, UNC93B deficiency in mouse models leads to increased mortality rate in mice that had been infected with HSV-1. Human studies also showed that UNC93B deficiency is a precipitating factor for HSV encephalitis. Polymorphisms of the UNC93B gene are associated with adult-onset HSE [21].
TRIF deficiency
Downstream signaling pathways after TLR stimulation are divided into two pathways: the first is the myeloid differentiation response 88 (MYD88) dependent pathway, and the second is the TRIF dependent pathway. TLR2, 5, 7, 8, 9 interact with MYD88. The TRIF pathway uses TLR3. Both pathways are important for inducing pro-inflammatory cytokines and IFN-producing genes [22]. TRIF is an adapter protein in the TLR3 and TLR4 signaling pathways. Autosomal recessive and autosomal dominant loss-of-function mutations have been reported in patients with HSE. Autosomal recessive TRIF deficiency is associated with a complete loss of function of TLR3 and TLR4-dependent pathways. But the autosomal dominant form is accompanied by partial TRIF deficiency with impairment of TLR3-mediated responses and intact TLR4-dependent pathways. Clinical penetrance in this form is incomplete [23-25]. TRIF-deficient fibroblasts do not produce IFN after stimulation and are susceptible to varicella-zoster virus and HSV infections [24].
Tumor necrosis factor receptor-associated factor (TRAF)-3 deficiency
TRAF3 is an important regulator of the immunologic system. The role of this adapter protein in human immunity is not well defined. TRAF3 is a cytoplasmic protein that mediates cellular signaling in some receptors, such as TLRs. It is a negative regulator of the (NF-KB) nuclear factor Kappa-light-chain-enhancer of activated B cells pathway. Autosomal dominant TRAF3 gene mutation is associated with HSE [26, 27]. Studies showed that an autosomal dominant de novo R118W germline mutation in TRAF3 may lead to HSE [28]. R118W variant is associated with low expression of TRAF3 protein and decreased production of Interleukin (IL)-6, INFβ/λ. This mutation was first described in a young adult with HSE in the childhood period [29].
TBK1 deficiency
TBK1 (TANK binding kinase 1) is an activator of the innate immune system. It contributes to signaling pathways that lead to the production of type 1 IFNs via NF-KB. Patients with TBK1 deficiency are prone to an immunocompromised state. TBK1 also has an important role in the early phases of autophagy. Selective autophagy has been reported in the clearance of Salmonella and Mycobacterium tuberculosis. In HSV-1 infection, autophagy also has a critical role. The interaction between HSV1-encoded products that inhibit host autophagy and TBK1-mediated autophagy plays an important role in the prevention of the dissemination of HSV1 in the CNS. Loss-of-function genetic defect in TBK1 can lead to HSE [30, 31]. Activation of TBK1 results in the phosphorylation of Interferon regulatory factor 3 (IRF3), leading to its homodimerization and subsequent translocation into the nucleus, where it stimulates the production of interferon (type 1, 3) [31]. Interferon has a critical role in controlling HSV infection during the disease.
Additionally, there is growing evidence for anti-viral effects of autophagy in herpes infections. Studies showed that early cytoplasmic autophagy can occur in HSV-negative cells adjacent to infected cells. Fibroblasts with TBK1 deficiency cannot stimulate this autophagy response in the field of HSV infection. Furthermore, it seems that early autophagy is an INF-independent process in HSV infection [32]. Jean-Laurent Casanova reported two unrelated children with forebrain HSE who had heterozygous mutations in the TBK1 gene. They had autosomal dominant inheritance. Loss-of-function alleles were reported in both mutations. However, they had different mechanisms. One mutation (D50A) leads to protein instability, and the second one (G159A) results in loss of kinase activity [7].
IRF3 deficiency
IRF3 is a signaling molecule that is activated by several pattern recognition receptors and contributes to the production of type 1 interferon. It is a critical factor in the innate immune response against viral infection. TBK1 phosphorylates IRF3 at Ser386 and ser396. Then, IRF3 dimerizes and translocates into the nucleus and induces interferon production [33]. An autosomal dominant loss-of-function mutation in the IRF3 gene may lead to HSE. R258Q mutation in IRF can fail phosphorylation and dimerization and finally impair the transcription process [34].
Small nucleolar RNA 31 (SNORA31) deficiency
SNORA31 is a 130-nucleotide snoRNA. The gene encoding snoRA31 is expressed in various human cell types. Pseudouridylation of uridine residue in position 218 of 18s ribosomal RNA (rRNA) and position 3713 of the 28s rRNA is the primary function of snoRA31. snoRA 31 is a critical factor in cell-intrinsic immunity to the herpes virus in the brain. Four heterozygous variants of SNORA31 in five unrelated families with HSE have been reported [3, 35].
Debranching enzyme 1 (DBR1) deficiency
DBR1 hydrolyzes intron lariat RNA to facilitate its processing. The level of this enzyme is very high in the brainstem and spinal cord. Casanova and colleagues found seven patients from three families with brainstem HSE, influenza B, and norovirus encephalitis that have a biallelic variant in the DBR1 gene. According to some studies, it seems that DBR1 has a critical role in controlling brainstem-intrinsic defenses against viral encephalitis by regulation of lariat metabolism [3, 36].
Signal transducer and activator of transcription 1 (STAT1) deficiency
STAT1 is a transcriptional factor that can be stimulated by several factors, such as interferon type 1, 2, 3, and IL-27. Four characteristic mutations with patterns of autosomal dominant and autosomal recessive inheritance have been reported. autosomal dominant STAT1 gain of function, autosomal dominant STAT1 deficiency, autosomal recessive partial STAT1 deficiency, and autosomal recessive complete STAT1 deficiency are genetic defects in the STAT1 gene with different clinical phenotypes [37]. STAT1 gain-of-function mutation can lead to impairment of natural killer (NK) cell proliferation and function. Furthermore, severe and recurrent cutaneous herpes simplex infection has been reported in patients with autosomal dominant gain-of-function STAT1 mutations. Recurrent herpes infection, disseminated varicella infection, fulminant Epstein-Barr virus infection, and HSE have been reported in patients with an autosomal recessive loss-of-function mutation in the STAT1 gene [38-40].
In summary, according to several studies, STAT1 variants based on the type of mutations and inheritance pattern have different clinical phenotypes. Autosomal dominant and autosomal recessive STAT1 loss of function mutation may present Mendelian susceptibility to mycobacterial disease, fungal infections, and HSE. Nevertheless, patients with autosomal dominant STAT1 gain-of-function mutation are prone to chronic mucocutaneous candidiasis, progressive multifocal leukoencephalopathy [41].
GTF3A gene mutation
Common variable immunodeficiency (CVID) is a humoral immunodeficiency disorder characterized by recurrent bacterial infections. These patients have immune dysregulation and autoimmunity. Patients with CVID have variable clinical manifestations. The most common infections are encapsulated bacterial infections. However, viral infections are infrequent in CVID. Few viral infections are reported in CVID, for example: chronic enteroviral meningoencephalitis, chronic enterovirus colitis, cytomegalovirus retinitis, recurrent enteroviral skin infections, and HSE. Ansari reported a case of CVID with HSE in 2010 [42-44]. Naesens and colleagues also report a compound heterogeneous loss-of-function mutation in the GFTIIIA gene in a Belgian family with CVID. Transcriptional factor IIIA is a component that has an important role in generating and stabilizing 5S rRNA. It is encoded by the GFTIIIA gene. This patient was admitted to the hospital because of HSE at the age of 9 months. After familial Sanger sequencing, they found that his sister has CVID. She was HSV-1 seropositive without any clinical presentation of severe HSV-1 infection [3, 45].
Conclusion
HSV is a neuroinvasive pathogen, and HSE is a rare presentation of HSV infection. Innate and acquired immunity have a central role in controlling HSV infection. There is incomplete evidence about the increased susceptibility to HSE in HSV-infected patients. The absence of HSE epidemics and the high frequency of consanguinity in parents of children with HSE highlight the possibility of a genetic basis. Studies strongly suggest that HSE has a genetic predisposition at least in some people. Several papers have shown that defects in TLR-3 signaling, IFN production, and IFN-independent cell-intrinsic mechanisms contribute to HSE in patients with HSV infection. Genetic defects in components of the TLR3 signaling pathway, such as TLR3, UNC93B1, TRIF, TRAF3, IRF3, and STAT1, have been reported as responsible genes for HSE. Other genetic mutations have also been found as the etiology of HSE in some people. Furthermore, despite HSE being a rare complication of HSV, genetic evaluation of these patients can diagnose some IEI and may give us new insights for future investigations in the fields of diagnosis, treatment, and prophylaxis.
Ethical Considerations
The Ethics Committee of Shahid Sadoughi University of Medical Sciences, Yazd, Iran, approved the study with the code No. IR.SSU.MEDICINE.REC.1396.1.
Funding Statement
None.
Conflict of Interest
The authors declared no conflict of interest.
Acknowledgments
None.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Authors' Contributions
A.A. was responsible for designing the review protocol, conducting the literature review, providing feedback on the manuscripts, writing the manuscript and improving the interpretation of the results. A.Kh. was responsible for writing the manuscript, assembling data, analyzing data, and interpreting analyses.
TRIF deficiency
Downstream signaling pathways after TLR stimulation are divided into two pathways: the first is the myeloid differentiation response 88 (MYD88) dependent pathway, and the second is the TRIF dependent pathway. TLR2, 5, 7, 8, 9 interact with MYD88. The TRIF pathway uses TLR3. Both pathways are important for inducing pro-inflammatory cytokines and IFN-producing genes [22]. TRIF is an adapter protein in the TLR3 and TLR4 signaling pathways. Autosomal recessive and autosomal dominant loss-of-function mutations have been reported in patients with HSE. Autosomal recessive TRIF deficiency is associated with a complete loss of function of TLR3 and TLR4-dependent pathways. But the autosomal dominant form is accompanied by partial TRIF deficiency with impairment of TLR3-mediated responses and intact TLR4-dependent pathways. Clinical penetrance in this form is incomplete [23-25]. TRIF-deficient fibroblasts do not produce IFN after stimulation and are susceptible to varicella-zoster virus and HSV infections [24].
Tumor necrosis factor receptor-associated factor (TRAF)-3 deficiency
TRAF3 is an important regulator of the immunologic system. The role of this adapter protein in human immunity is not well defined. TRAF3 is a cytoplasmic protein that mediates cellular signaling in some receptors, such as TLRs. It is a negative regulator of the (NF-KB) nuclear factor Kappa-light-chain-enhancer of activated B cells pathway. Autosomal dominant TRAF3 gene mutation is associated with HSE [26, 27]. Studies showed that an autosomal dominant de novo R118W germline mutation in TRAF3 may lead to HSE [28]. R118W variant is associated with low expression of TRAF3 protein and decreased production of Interleukin (IL)-6, INFβ/λ. This mutation was first described in a young adult with HSE in the childhood period [29].
TBK1 deficiency
TBK1 (TANK binding kinase 1) is an activator of the innate immune system. It contributes to signaling pathways that lead to the production of type 1 IFNs via NF-KB. Patients with TBK1 deficiency are prone to an immunocompromised state. TBK1 also has an important role in the early phases of autophagy. Selective autophagy has been reported in the clearance of Salmonella and Mycobacterium tuberculosis. In HSV-1 infection, autophagy also has a critical role. The interaction between HSV1-encoded products that inhibit host autophagy and TBK1-mediated autophagy plays an important role in the prevention of the dissemination of HSV1 in the CNS. Loss-of-function genetic defect in TBK1 can lead to HSE [30, 31]. Activation of TBK1 results in the phosphorylation of Interferon regulatory factor 3 (IRF3), leading to its homodimerization and subsequent translocation into the nucleus, where it stimulates the production of interferon (type 1, 3) [31]. Interferon has a critical role in controlling HSV infection during the disease.
Additionally, there is growing evidence for anti-viral effects of autophagy in herpes infections. Studies showed that early cytoplasmic autophagy can occur in HSV-negative cells adjacent to infected cells. Fibroblasts with TBK1 deficiency cannot stimulate this autophagy response in the field of HSV infection. Furthermore, it seems that early autophagy is an INF-independent process in HSV infection [32]. Jean-Laurent Casanova reported two unrelated children with forebrain HSE who had heterozygous mutations in the TBK1 gene. They had autosomal dominant inheritance. Loss-of-function alleles were reported in both mutations. However, they had different mechanisms. One mutation (D50A) leads to protein instability, and the second one (G159A) results in loss of kinase activity [7].
IRF3 deficiency
IRF3 is a signaling molecule that is activated by several pattern recognition receptors and contributes to the production of type 1 interferon. It is a critical factor in the innate immune response against viral infection. TBK1 phosphorylates IRF3 at Ser386 and ser396. Then, IRF3 dimerizes and translocates into the nucleus and induces interferon production [33]. An autosomal dominant loss-of-function mutation in the IRF3 gene may lead to HSE. R258Q mutation in IRF can fail phosphorylation and dimerization and finally impair the transcription process [34].
Small nucleolar RNA 31 (SNORA31) deficiency
SNORA31 is a 130-nucleotide snoRNA. The gene encoding snoRA31 is expressed in various human cell types. Pseudouridylation of uridine residue in position 218 of 18s ribosomal RNA (rRNA) and position 3713 of the 28s rRNA is the primary function of snoRA31. snoRA 31 is a critical factor in cell-intrinsic immunity to the herpes virus in the brain. Four heterozygous variants of SNORA31 in five unrelated families with HSE have been reported [3, 35].
Debranching enzyme 1 (DBR1) deficiency
DBR1 hydrolyzes intron lariat RNA to facilitate its processing. The level of this enzyme is very high in the brainstem and spinal cord. Casanova and colleagues found seven patients from three families with brainstem HSE, influenza B, and norovirus encephalitis that have a biallelic variant in the DBR1 gene. According to some studies, it seems that DBR1 has a critical role in controlling brainstem-intrinsic defenses against viral encephalitis by regulation of lariat metabolism [3, 36].
Signal transducer and activator of transcription 1 (STAT1) deficiency
STAT1 is a transcriptional factor that can be stimulated by several factors, such as interferon type 1, 2, 3, and IL-27. Four characteristic mutations with patterns of autosomal dominant and autosomal recessive inheritance have been reported. autosomal dominant STAT1 gain of function, autosomal dominant STAT1 deficiency, autosomal recessive partial STAT1 deficiency, and autosomal recessive complete STAT1 deficiency are genetic defects in the STAT1 gene with different clinical phenotypes [37]. STAT1 gain-of-function mutation can lead to impairment of natural killer (NK) cell proliferation and function. Furthermore, severe and recurrent cutaneous herpes simplex infection has been reported in patients with autosomal dominant gain-of-function STAT1 mutations. Recurrent herpes infection, disseminated varicella infection, fulminant Epstein-Barr virus infection, and HSE have been reported in patients with an autosomal recessive loss-of-function mutation in the STAT1 gene [38-40].
In summary, according to several studies, STAT1 variants based on the type of mutations and inheritance pattern have different clinical phenotypes. Autosomal dominant and autosomal recessive STAT1 loss of function mutation may present Mendelian susceptibility to mycobacterial disease, fungal infections, and HSE. Nevertheless, patients with autosomal dominant STAT1 gain-of-function mutation are prone to chronic mucocutaneous candidiasis, progressive multifocal leukoencephalopathy [41].
GTF3A gene mutation
Common variable immunodeficiency (CVID) is a humoral immunodeficiency disorder characterized by recurrent bacterial infections. These patients have immune dysregulation and autoimmunity. Patients with CVID have variable clinical manifestations. The most common infections are encapsulated bacterial infections. However, viral infections are infrequent in CVID. Few viral infections are reported in CVID, for example: chronic enteroviral meningoencephalitis, chronic enterovirus colitis, cytomegalovirus retinitis, recurrent enteroviral skin infections, and HSE. Ansari reported a case of CVID with HSE in 2010 [42-44]. Naesens and colleagues also report a compound heterogeneous loss-of-function mutation in the GFTIIIA gene in a Belgian family with CVID. Transcriptional factor IIIA is a component that has an important role in generating and stabilizing 5S rRNA. It is encoded by the GFTIIIA gene. This patient was admitted to the hospital because of HSE at the age of 9 months. After familial Sanger sequencing, they found that his sister has CVID. She was HSV-1 seropositive without any clinical presentation of severe HSV-1 infection [3, 45].
Conclusion
HSV is a neuroinvasive pathogen, and HSE is a rare presentation of HSV infection. Innate and acquired immunity have a central role in controlling HSV infection. There is incomplete evidence about the increased susceptibility to HSE in HSV-infected patients. The absence of HSE epidemics and the high frequency of consanguinity in parents of children with HSE highlight the possibility of a genetic basis. Studies strongly suggest that HSE has a genetic predisposition at least in some people. Several papers have shown that defects in TLR-3 signaling, IFN production, and IFN-independent cell-intrinsic mechanisms contribute to HSE in patients with HSV infection. Genetic defects in components of the TLR3 signaling pathway, such as TLR3, UNC93B1, TRIF, TRAF3, IRF3, and STAT1, have been reported as responsible genes for HSE. Other genetic mutations have also been found as the etiology of HSE in some people. Furthermore, despite HSE being a rare complication of HSV, genetic evaluation of these patients can diagnose some IEI and may give us new insights for future investigations in the fields of diagnosis, treatment, and prophylaxis.
Ethical Considerations
The Ethics Committee of Shahid Sadoughi University of Medical Sciences, Yazd, Iran, approved the study with the code No. IR.SSU.MEDICINE.REC.1396.1.
Funding Statement
None.
Conflict of Interest
The authors declared no conflict of interest.
Acknowledgments
None.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Authors' Contributions
A.A. was responsible for designing the review protocol, conducting the literature review, providing feedback on the manuscripts, writing the manuscript and improving the interpretation of the results. A.Kh. was responsible for writing the manuscript, assembling data, analyzing data, and interpreting analyses.
References
- Khalili A. A review of primary immuno-deficiency disorders predisposing to cancer. Journal of Shahid Sadoughi University of Medical Sciences 2024; 31(11): 7179-193.
- Rozenberg F. [Herpes simplex virus and central nervous system infections: Encephalitis, meningitis, myelitis]. Virologie (Montrouge, France). 2020; 24(5): 283-94.
- Skouboe MK, Werner M. Inborn errors of immunity predisposing to herpes simplex virus infections of the central nervous system. Pathogens 2023; 12(2): 310.
- Costa BKD, Sato DK. Viral encephalitis: a practical review on diagnostic approach and treatment. J pediatr. 2020; 96(S 1): 12-9.
- Zhu S, Viejo-Borbolla A. Pathogenesis and virulence of herpes simplex virus. Virulence 2021; 12(1): 2670-702.
- Messacar K, Fischer M, Dominguez SR, Tyler KL, Abzug MJ. Encephalitis in US Children. Infectious Disease Clinics of North America 2018; 32(1): 145-62.
- Zhang SY. Herpes simplex virus encephalitis of childhood: inborn errors of central nervous system cell-intrinsic immunity. Human Genetics 2020; 139(6-7): 911-18.
- Kennedy PG. An overview of viral infections of the nervous system in the immunosuppressed. Journal of Neurology 2021; 268(8): 3026-3030.
- Duan T, Du Y, Xing C, Wang HY, Wang RF. Toll-like receptor signaling and its role in cell-mediated immunity. Frontiers in Immunology 2022; 13: 812774.
- Zheng W, Xu Q, Zhang Y, E X, Gao W, Zhang M, et al. Toll-like receptor-mediated innate immunity against herpesviridae infection: A current perspective on viral infection signaling pathways. Virology Journal 2020; 17: 1-15.
- Yujuan C, Junhong L, Yao Z, Xianping M, Huashan Y. Toll-like receptor 3 (TLR3) regulation mechanisms and roles in antiviral innate immune responses. Journal of Zhejiang University Science 2021; 22(8): 609.
- Mielcarska MB, Bossowska-Nowicka M, Toka FN. Functional failure of TLR3 and its signaling components contribute to herpes simplex encephalitis. J Neuroimmunol. 2018; 316: 65-73.
- Cleaver J, Jeffery K, Klenerman P, Lim M, Handunnetthi L, Irani SR, et al. The immunobiology of herpes simplex virus encephalitis and post-viral autoimmunity. Brain 2023; 2(1): 419.
- Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. Journal of Experimental Medicine 2011; 208(10): 2083-98.
- Mielcarska MB, Bossowska-Nowicka M, Toka FN. Functional failure of TLR3 and its signaling components contribute to herpes simplex encephalitis. Journal of Neuroimmunology 2018; 316: 65-73.
- Armangue T, Baucells BJ, Vlagea A, Petit-Pedrol M, Esteve-Solé A, Deyà-Martínez A, et al. Toll-like receptor 3 deficiency in autoimmune encephalitis post–herpes simplex encephalitis. Neurology: Neuroimmunology & Neuroinflammation 2019; 6(6): 611.
- Piret J, Boivin G. Innate immune response during herpes simplex virus encephalitis and development of immunomodulatory strategies. Reviews in medical virology 2015; 25(5): 300-19.
- Zhang L, Li F, Liu W, Tai Z, Yang J, et al. When herpes simplex virus encephalitis meets antiviral innate immunity. Frontiers in Immunology 2023; 14: 1118236.
- Pelka K, Bertheloot D, Reimer E, Phulphagar K, Schmidt SV, Christ A, et al. The chaperone UNC93B1 regulates Toll-like receptor stability independently of endosomal TLR transport. Immunity 2018; 48(5): 911-22.
- Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochemical and Biophysical Research Communications 2009; 388(4): 621-25.
- Wang JP, Bowen GN, Zhou S, Cerny A, Zacharia A, Knipe DM, et al. Role of specific innate immune responses in herpes simplex virus infection of the central nervous system. Journal of Virology 2012; 86(4): 2273-281.
- Kim AY, Shim HJ, Kim SY, Heo S, Youn HS. Differential regulation of MyD88-and TRIF-dependent signaling pathways of Toll-like receptors by cardamonin. International Immunopharmacology 2018; 64: 1-9.
- van der Made CI, Hoischen A, Netea MG, van de Veerdonk FL. Primary immunodeficiencies in cytosolic pattern‐recognition receptor pathways: Toward host‐directed treatment strategies. Immunological Reviews 2020; 297(1): 247-72.
- Sancho-Shimizu V, de Diego RP, Jouanguy E, Zhang SY, Casanova JL. Inborn errors of anti-viral interferon immunity in humans. Current Opinion in Virology 2011; 1(6): 487-96.
- Menasria R, Boivin N, Lebel M, Piret J, Gosselin J, Boivin G. Both TRIF and IPS-1 adaptor proteins contribute to the cerebral innate immune response against herpes simplex virus 1 infection. Journal of Virology 2013; 87(13): 7301-738.
- Rae W, Sowerby JM, Verhoeven D, Youssef M, Kotagiri P, Savinykh N, et al. Immunodeficiency, autoimmunity, and increased risk of B cell malignancy in humans with TRAF3 mutations. Science Immunology 2022; 7(74): 3800.
- Alsweed A, Alsuhibani M, Casanova JL, Al-Hajjar S. Approach to recurrent herpes simplex encephalitis in children. International Journal of Pediatrics and Adolescent Medicine 2018; 5(2): 35-8.
- de Diego RP, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 2010; 33(3): 400-11.
- Aluri J, Cooper MA, Schuettpelz LG. Toll-like receptor signaling in the establishment and function of the immune system. Cells 2021; 10(6): 1374.
- Taft J, Markson M, Legarda D, Patel R, Chan M, Malle L, et al. Human TBK1 deficiency leads to autoinflammation driven by TNF-induced cell death. Cell 2021; 184(17): 4447-463.
- Ahmad L, Zhang SY, Casanova JL, Sancho-Shimizu V. Human TBK1: a gatekeeper of neuroinflammation. Trends in Molecular Medicine 2016; 22(6): 511-27.
- Liyana A, Vanessa SS. The emerging role of human TBK1 in virus-induced autophagy. Autophagy 2019; 15(5): 917-18.
- Wang J, Li H, Xue B, Deng R, Huang X, Xu Y, et al. IRF1 promotes the innate immune response to viral infection by enhancing the activation of IRF3. Journal of Virology 2020; 94(22): 1220-231.
- Andersen LL, Mørk N, Reinert LS, Kofod-Olsen E, Narita R, Jørgensen SE, et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. Journal of Experimental Medicine 2015; 212(9): 1371-379.
- Lafaille FG, Harschnitz O, Lee YS, Zhang P, Hasek ML, Kerner G, et al. Human SNORA31 variations impair cortical neuron-intrinsic immunity to HSV-1 and underlie herpes simplex encephalitis. Nature Medicine 2019; 25(12): 1873-884.
- Skouboe MK, Werner M, Mogensen TH. Inborn errors of immunity predisposing to herpes simplex virus infections of the central nervous system. Pathogens 2023; 12(2): 310.
- Mizoguchi Y, Okada S. Inborn errors of STAT1 immunity. Current Opinion in Immunology 2021; 72(1): 59-64.
- Jouanguy E, Béziat V, Mogensen TH, Casanova JL, Tangye SG, Zhang SY. Human inborn errors of immunity to herpes viruses. Current Opinion in Immunology 2020; 62: 106-22.
- Khalili A. A Review of Primary Immunodeficiency Diseases with Skin Manifestations. Journal of Shahid Sadoughi University of Medical Sciences 2022; 29(10): 4164-179.
- Erdős M, Jakobicz E, Soltész B, Tóth B, Bata-Csörgő Z, Maródi L. Recurrent, severe aphthous stomatitis and mucosal ulcers as primary manifestations of a novel stat1 gain-of-function mutation. Frontiers in Immunology 2020; 11: 967.
- Mogensen TH. IRF and STAT transcription factors-from basic biology to roles in infection, protective immunity, and primary immunodeficiencies. Frontiers in Immunology 2019; 9: 3047.
- Asgardoon MH, Azizi G, Yazdani R, Sohani M, Pashangzadeh S, Kalantari A, et al. Monogenic primary immunodeficiency disorder associated with common variable immunodeficiency and autoimmunity. International Archives of Allergy and Immunology 2020; 181(9): 706-14.
- Ansari M, Jaha S. Herpes simplex encephalitis in a patient having common variable immuno-deficiency. Annals of Tropical Medicine and Public Health 2010; 3(1): 30.
- Khalili A. Efficacy of ganciclovir on cmv retinitis complication of common variable immunodeficiency. Immunology and Genetics Journal 2018; 1: 103-107.
- Naesens L, Muppala S, Acharya D, Nemegeer J, Bogaert D, Lee JH, et al. GTF3A mutations predispose to herpes simplex encephalitis by disrupting biogenesis of the host-derived RIG-I ligand RNA5SP141. Science Immunology 2022; 7(77): 4531.
Type of Study: Research |
Subject:
Virology
Received: 2025/05/28 | Accepted: 2024/08/31 | Published: 2024/10/31
Received: 2025/05/28 | Accepted: 2024/08/31 | Published: 2024/10/31
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
